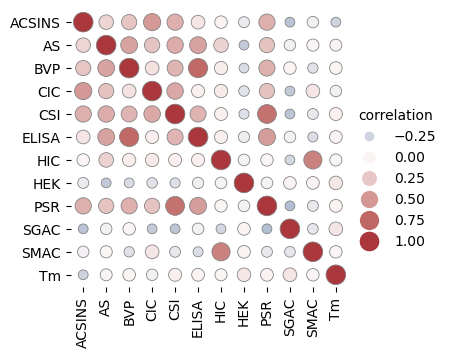
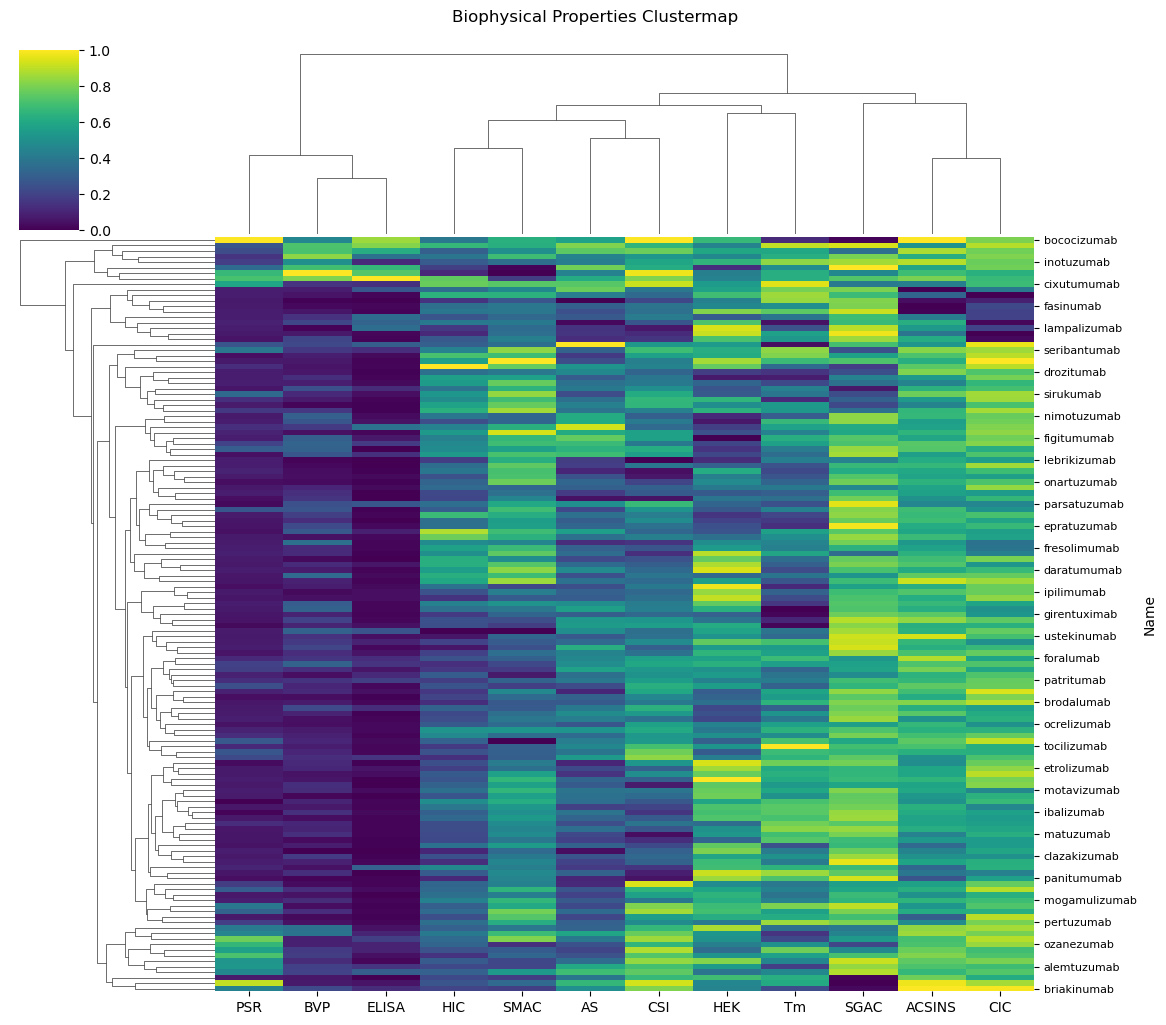
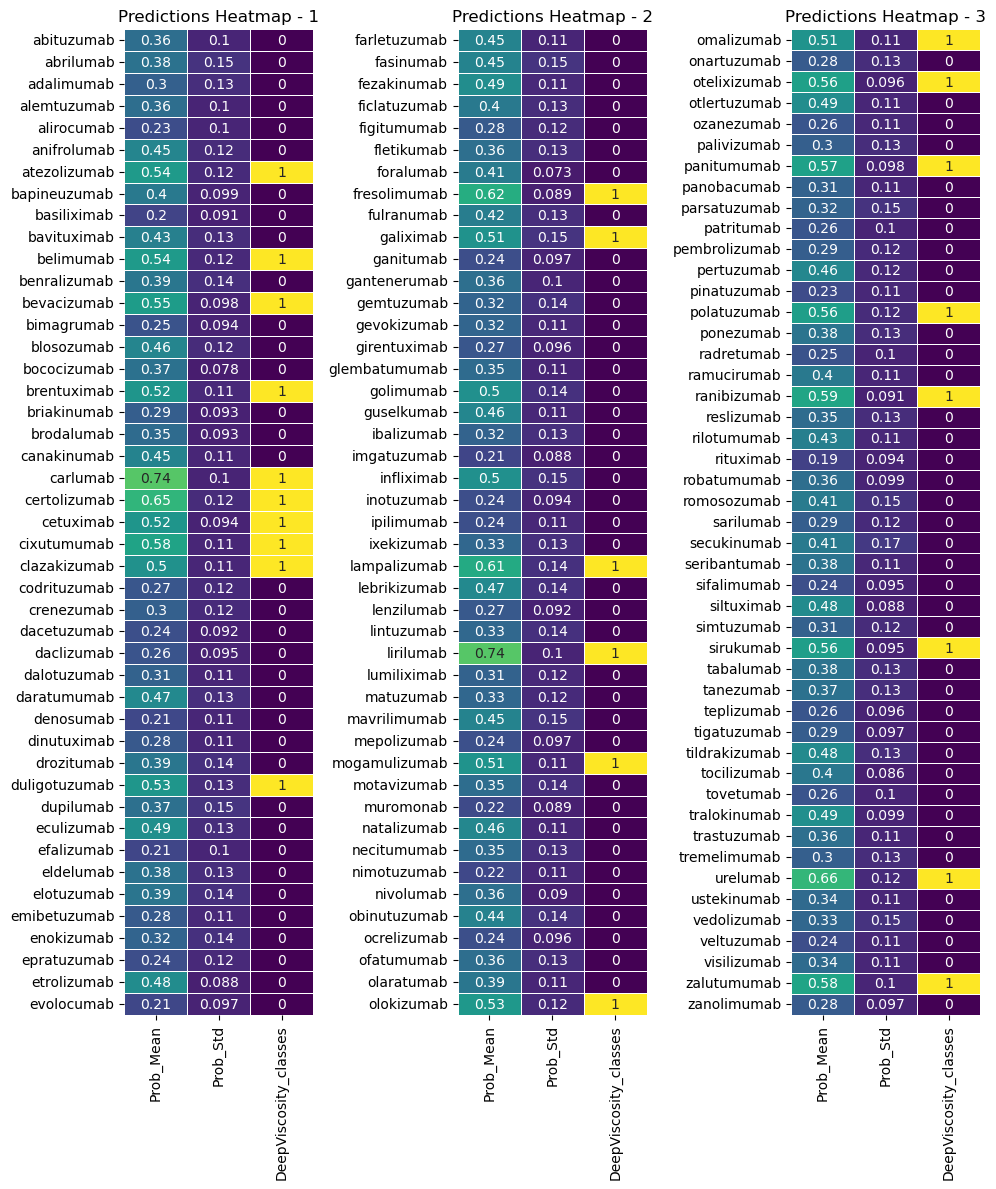
import os
import joblib
import numpy as np
import pandas as pd
from sklearn.preprocessing import StandardScaler
def predict_biophysical_properties(data_path: str, models_dir: str) -> pd.DataFrame:
"""
CSV 파일을 읽어와서 여러 모델에 필요한 특징을 전처리하고, 사전 훈련된 모델을 불러와 예측을 수행한 후, 그 결과를 CSV 파일로 저장합니다.
* `data_path` (str): 입력 CSV 파일 경로 (예: 'SAPSCM.csv').
* `models_dir` (str): 훈련된 모델의 joblib 파일이 있는 디렉터리.
Returns):
`pd.DataFrame`: 각 샘플에 대한 예측된 생물물리학적 특성을 포함하는 데이터프레임. 오류가 발생하면 빈 데이터프레임을 반환합니다.
"""
try:
# Load the input data
df = pd.read_csv(data_path)
except FileNotFoundError:
print(f"Error: The file at {data_path} was not found.")
return pd.DataFrame()
except Exception as e:
print(f"An error occurred while loading the data: {e}")
return pd.DataFrame()
# Define the features and their corresponding model types and filenames
prediction_configs = {
"ACSINS": {
"features": [
"SAP_pos_CDRH2",
"SAP_pos_CDRL3",
"SCM_pos_CDRH1",
"SCM_neg_CDR",
],
"model_type": "SVR",
},
"AS": {
"features": [
"SAP_pos_CDRH3",
"SCM_pos_CDRL2",
"SCM_pos_CDRL3",
"SCM_neg_CDRL3",
],
"model_type": "LR",
},
"BVP": {
"features": [
"SAP_pos_CDRH2",
"SAP_pos_CDRH3",
"SCM_pos_CDR",
"SCM_neg_CDRH3",
],
"model_type": "KNN",
},
"CIC": {
"features": ["SAP_pos_CDRL3", "SAP_pos_CDRL3", "SAP_pos_Lv", "SCM_neg_CDR"],
"model_type": "KNN",
},
"CSI": {
"features": [
"SAP_pos_CDRL1",
"SAP_pos_Lv",
"SCM_pos_CDRH2",
"SCM_neg_CDRL2",
],
"model_type": "SVR",
},
"ELISA": {
"features": ["SAP_pos_CDRH3", "SCM_pos_CDR", "SCM_neg_CDR"],
"model_type": "KNN",
},
"HIC": {
"features": ["SAP_pos_CDRL3", "SAP_pos_CDR", "SAP_pos_Hv", "SCM_pos_CDRH3"],
"model_type": "SVR",
},
"HEK": {
"features": ["SAP_pos_CDRH2", "SAP_pos_CDRL3", "SCM_pos_Lv", "SCM_neg_Lv"],
"model_type": "KNN",
},
"PSR": {
"features": ["SAP_pos_Lv", "SCM_pos_CDRH2", "SCM_neg_CDRL2"],
"model_type": "SVR",
},
"SGAC": {
"features": [
"SAP_pos_CDRH1",
"SAP_pos_CDRL3",
"SCM_neg_CDRH2",
"SCM_neg_Lv",
],
"model_type": "SVR",
},
"SMAC": {
"features": ["SAP_pos_CDR", "SAP_pos_Fv", "SCM_neg_CDRL2", "SCM_neg_Fv"],
"model_type": "KNN",
},
"Tm": {
"features": ["SAP_pos_CDRH1", "SAP_pos_CDRH2", "SCM_pos_CDRH3"],
"model_type": "KNN",
},
}
sc = StandardScaler()
results = {}
for prop_name, config in prediction_configs.items():
features = config["features"]
model_type = config["model_type"]
# Select features from the DataFrame and transform them
try:
X = df[features].values
# Check if features exist to avoid KeyError
if X.shape[1] != len(features):
print(f"Warning: Not all features for {prop_name} were found. Skipping.")
continue
# Transform the feature data
X_scaled = sc.fit_transform(X)
# Construct the model file path
model_file = f"{prop_name}_{model_type}_model.joblib"
model_path = os.path.join(models_dir, model_file)
# Load the model
model = joblib.load(model_path)
# Make a prediction and store the result
results[prop_name] = model.predict(X_scaled)
except KeyError as e:
print(f"Error: Missing columns for property {prop_name}. Columns: {e}")
continue
except FileNotFoundError:
print(f"Error: Model file not found at {model_path}. Skipping.")
continue
except Exception as e:
print(f"An unexpected error occurred for {prop_name}: {e}")
continue
if not results:
print("No predictions were successfully made. Returning empty DataFrame.")
return pd.DataFrame()
# Combine all results into a single DataFrame
result_df = pd.DataFrame(results, index=df["Name"])
result_df.index.name = "Name"
result_df.reset_index(inplace=True)
return result_df
# Define paths
PATH = "../data/example/"
MODELS_DIR = "../models/Trained_model"
# Load the data and predict properties
results_df = predict_biophysical_properties(os.path.join(PATH, "SAPSCM.csv"), MODELS_DIR)
if not results_df.empty:
# Save the results to a new CSV file
output_path = os.path.join(PATH, "AbDev_result.csv")
results_df.to_csv(output_path, index=False)
# Display the first few rows of the final DataFrame
print(results_df.head())