Rosalind Stronghold 문제풀이

생물정보학의 다양한 주제인 질량 분석, 서열 정렬, 동적 프로그래밍, 게놈 어셈블리, 계통 발생, 확률, 문자열 알고리즘 등의 기초가 되는 알고리즘에 대해 알아봅니다.
Rosalind 는 프로젝트 오일러, 구글 코드 잼에서 영감을 얻었습니다. 이 프로젝트의 이름은 DNA 이중나선을 발견하는 데 기여한 로잘린드 프랭클린 에서 따왔습니다. Rosalind 는 프로그래밍 실력을 키우고자 하는 생물학자와 분자생물학의 계산 문제를 접해본 적이 없는 프로그래머들에게 도움이 될 것입니다.
1 Counting DNA Nucleotides
문자열 은 단순히 어떤 알파벳 에서 선택되어 단어로 구성된 기호의 정렬된 모음이며, 문자열의 길이 는 문자열에 포함된 기호의 수입니다.
길이 21 의 DNA 문자열 의 예 (알파벳에 ‘A’, ‘C’, ‘G’, ‘T’ 기호가 포함됨) 는 “ATGCTTCAGAAAGGTCTTACG” 입니다.
Given: 최대 1000nt 길이의 DNA 문자열 s 입니다.
Return: 반환: s 에서 ‘A’, ‘C’, ‘G’, ‘T’ 기호가 각각 나타나는 횟수를 세는 4 개의 정수 (공백으로 구분) 를 반환합니다.
1.1 Sample Dataset
AGCTTTTCATTCTGACTGCAACGGGCAATATGTCTCTGTGTGGATTAAAAAAAGAGTGTCTGATAGCAGC1.2 Sample Output
20 12 17 211.3 Solution
주어진 DNA 문자열 ‘s’ 에서 각 뉴클레오티드 (‘A’, ‘C’, ‘G’, ‘T’) 의 발생 횟수를 세는 문제를 해결하려면 다음 단계를 따르세요.
- 카운터를 초기화합니다: ‘A’, ‘C’, ‘G’, ‘T’ 에 대한 카운터를 설정합니다.
- 문자열을 반복합니다: 문자열의 각 문자를 순회하며 해당 카운터를 증가시킵니다.
- 결과를 출력합니다: ‘A’, ‘C’, ‘G’, ‘T’ 의 개수를 공백으로 구분하여 인쇄합니다.
def count_nucleotides(dna_string):
count_A = 0
count_C = 0
count_G = 0
count_T = 0
for nucleotide in dna_string:
if nucleotide == 'A':
count_A += 1
elif nucleotide == 'C':
count_C += 1
elif nucleotide == 'G':
count_G += 1
elif nucleotide == 'T':
count_T += 1
return count_A, count_C, count_G, count_T
# Sample Dataset
s = "AGCTTTTCATTCTGACTGCAACGGGCAATATGTCTCTGTGTGGATTAAAAAAAGAGTGTCTGATAGCAGC"
result = count_nucleotides(s)
print(" ".join(map(str, result)))
# Output should be "20 12 17 21"1.4 설명
- 초기화: ‘A’, ‘C’, ‘G’, ‘T’ 의 카운터가 0 으로 초기화됩니다.
- 각 문자를 반복합니다: 루프는 DNA 문자열의 각 문자를 검사하고 발견된 문자에 따라 해당 카운터를 증가시킵니다.
- 결과를 반환하고 인쇄합니다: 이 함수는 카운트를 반환한 다음 필요한 형식으로 출력합니다.
이 접근 방식은 각 뉴클레오타이드가 문자열을 한 번 통과할 때 효율적으로 카운트되도록 보장하며, 시간 복잡도는 \(O(n)\) 입니다.
2 Transcribing DNA into RNA
RNA 문자열은 ‘A’, ‘C’, ‘G’, ‘U’ 가 포함된 알파벳으로 구성된 문자열입니다.
코딩 가닥에 해당하는 DNA 문자열 t 가 주어지면, t 의 모든 ‘T’ 를 u 의 ‘U’ 로 대체하여 전사된 RNA 문자열 u 가 형성됩니다.
주어진: 길이가 최대 1000 nt 인 DNA 문자열 t 가 주어집니다. 반환합니다: 반환: t 의 전사된 RNA 문자열.
2.1 Sample Dataset
GATGGAACTTGACTACGTAAATT2.2 Sample Output
GAUGGAACUUGACUACGUAAAUU2.3 Solution
To transcribe a DNA string to an RNA string, we need to replace every occurrence of the nucleotide ‘T’ in the DNA string with ‘U’ to form the RNA string. This is because RNA uses uracil (U) instead of thymine (T).
def transcribe_dna_to_rna(dna_string):
# Replace all occurrences of 'T' with 'U'
rna_string = dna_string.replace('T', 'U')
return rna_string
# Sample Dataset
dna_string = "GATGGAACTTGACTACGTAAATT"
print(transcribe_dna_to_rna(dna_string)) # Output should be "GAUGGAACUUGACUACGUAAAUU"2.4 Explanation
- Function Definition:
transcribe_dna_to_rna(dna_string)takes a DNA string as input. - String Replacement:
dna_string.replace('T', 'U')creates a new string where allTs are replaced withUs. - Return Statement: The resulting RNA string is returned.
3 Complementing a Strand of DNA
In DNA strings, symbols ‘A’ and ‘T’ are complements of each other, as are ‘C’ and ‘G’.
The reverse complement of a DNA string s is the string sc𝑠c formed by reversing the symbols of s𝑠, then taking the complement of each symbol (e.g., the reverse complement of “GTCA” is “TGAC”).
Given: A DNA string s of length at most 1000 bp.
Return: The reverse complement sc𝑠c of s𝑠.
3.1 Sample Dataset
AAAACCCGGT3.2 Sample Output
ACCGGGTTTT3.3 soultion
To find the reverse complement of a DNA string, follow these steps:
- Reverse the string: First, reverse the input DNA string.
- Complement the string: Replace each nucleotide with its complement: ‘A’ with ‘T’, ‘T’ with ‘A’, ‘C’ with ‘G’, and ‘G’ with ‘C’.
def reverse_complement(dna_string):
# Dictionary to map each nucleotide to its complement
complement = {'A': 'T', 'T': 'A', 'C': 'G', 'G': 'C'}
# Reverse the DNA string
reversed_dna = dna_string[::-1]
# Replace each nucleotide with its complement
reverse_complement_dna = ''.join(complement[base] for base in reversed_dna)
return reverse_complement_dna
# Sample Dataset
dna_string = "AAAACCCGGT"
print(reverse_complement(dna_string))
# Output should be "ACCGGGTTTT"3.4 Explanation
- Complement Mapping:
- A dictionary
complementis used to map each nucleotide to its complementary nucleotide.
- A dictionary
- Reversing the String:
- The slicing operation
dna_string[::-1]reverses the string.
- The slicing operation
- Generating the Complement:
- A list comprehension is used to replace each nucleotide in the reversed string with its complement.
''.join()combines the list of complemented nucleotides into a single string.
4 Rabbits and Recurrence Relations
A sequence is an ordered collection of objects (usually numbers), which are allowed to repeat. Sequences can be finite or infinite. Two examples are the finite sequence and the infinite sequence of odd numbers \((1,3,5,7,9,…)\). We use the notation an𝑎𝑛 to represent the n-th term of a sequence.
A recurrence relation is a way of defining the terms of a sequence with respect to the values of previous terms. In the case of Fibonacci’s rabbits from the introduction, any given month will contain the rabbits that were alive the previous month, plus any new offspring. A key observation is that the number of offspring in any month is equal to the number of rabbits that were alive two months prior. As a result, if \(Fn\) represents the number of rabbit pairs alive after the n-th month, then we obtain the Fibonacci sequence having terms \(Fn\) that are defined by the recurrence relation \(Fn=Fn−1+Fn−2Fn=F_(n-1)+F_(n-2)\) (with \(F1=F2=1\) to initiate the sequence). Although the sequence bears Fibonacci’s name, it was known to Indian mathematicians over two millennia ago.
When finding the \(n\)-th term of a sequence defined by a recurrence relation, we can simply use the recurrence relation to generate terms for progressively larger values of n𝑛. This problem introduces us to the computational technique of dynamic programming, which successively builds up solutions by using the answers to smaller cases.
Given: Positive integers \(n≤40\) and \(k≤5\).
Return: The total number of rabbit pairs that will be present after n months, if we begin with 1 pair and in each generation, every pair of reproduction-age rabbits produces a litter of \(k\) rabbit pairs (instead of only 1 pair).
4.1 Sample Dataset
5 34.2 Sample Output
194.3 Solution
To solve the problem of computing the total number of rabbit pairs after a given number of months \(n\) when each pair of reproduction-age rabbits produces \(k\) rabbit pairs each month, we can modify the classic Fibonacci sequence. Instead of each rabbit pair producing just one new pair, they produce \(k\) new pairs.
Let’s break down the steps to create the solution:
Define the recurrence relation: The problem can be modeled with a recurrence relation. Let \(F(n)\) represent the number of rabbit pairs after \(n\) months. The recurrence relation can be expressed as: \[ F(n) = F(n-1) + k \times F(n-2) \] Here, \(F(n-1)\) represents the number of rabbit pairs from the previous month, and \(k\) times \(F(n-2)\) represents the new rabbit pairs produced by the pairs from two months ago.
Initial conditions:
- \(F(1) = 1\) (initially, there is one pair of rabbits)
- \(F(2) = 1\) (in the second month, there is still only one pair, as they have not yet reproduced)
Iterative computation: Using a loop, compute the number of rabbit pairs for each month up to \(n\) based on the recurrence relation.
Here is the Python function to implement this approach:
def total_rabbit_pairs(n, k):
if n == 1 or n == 2:
return 1
# Initialize the first two months
F1 = 1
F2 = 1
# Compute the number of rabbit pairs for each subsequent month
for month in range(3, n + 1):
F_current = F2 + k * F1
F1 = F2
F2 = F_current
return F2
# Sample Dataset
n = 5
k = 3
print(total_rabbit_pairs(n, k)) # Output should be 194.4 Explanation of the Code
- Base Cases:
- If \(n\) is 1 or 2, the function returns 1 because the first two terms are both 1.
- Initialization:
- Variables
F1andF2are initialized to 1, representing the number of rabbit pairs in the first and second months, respectively.
- Variables
- Loop Through Months:
- For each month from 3 to \(n\) , the number of rabbit pairs is calculated using the recurrence relation.
F_currentis calculated as the sum of the number of rabbit pairs from the previous month (F2) and the number of new rabbit pairs produced by the pairs from two months ago (k * F1).
- For each month from 3 to \(n\) , the number of rabbit pairs is calculated using the recurrence relation.
- Update Variables:
- After computing
F_current, updateF1andF2to the values of the last two computed terms to prepare for the next iteration.
- After computing
- Return the Result:
- Finally, return
F2, which holds the number of rabbit pairs after \(n\) months.
- Finally, return
5 Computing GC Content
The GC-content of a DNA string is given by the percentage of symbols in the string that are ‘C’ or ‘G’. For example, the GC-content of “AGCTATAG” is 37.5%. Note that the reverse complement of any DNA string has the same GC-content.
DNA strings must be labeled when they are consolidated into a database. A commonly used method of string labeling is called FASTA format. In this format, the string is introduced by a line that begins with ‘>’, followed by some labeling information. Subsequent lines contain the string itself; the first line to begin with ‘>’ indicates the label of the next string.
In Rosalind’s implementation, a string in FASTA format will be labeled by the ID “Rosalind_xxxx”, where “xxxx” denotes a four-digit code between 0000 and 9999.
Given: At most 10 DNA strings in FASTA format (of length at most 1 kbp each).
Return: The ID of the string having the highest GC-content, followed by the GC-content of that string. Rosalind allows for a default error of 0.001 in all decimal answers unles otherwise stated; please see the note on absolute error below.
5.1 Sample Dataset
>Rosalind_6404
CCTGCGGAAGATCGGCACTAGAATAGCCAGAACCGTTTCTCTGAGGCTTCCGGCCTTCCC
TCCCACTAATAATTCTGAGG
>Rosalind_5959
CCATCGGTAGCGCATCCTTAGTCCAATTAAGTCCCTATCCAGGCGCTCCGCCGAAGGTCT
ATATCCATTTGTCAGCAGACACGC
>Rosalind_0808
CCACCCTCGTGGTATGGCTAGGCATTCAGGAACCGGAGAACGCTTCAGACCAGCCCGGAC
TGGGAACCTGCGGGCAGTAGGTGGAAT5.2 Sample Output
Rosalind_0808
60.9195405.3 Solution
To solve this problem, we need to compute the GC-content of multiple DNA strings provided in FASTA format and identify the string with the highest GC-content.
5.4 Steps to Solve the Problem
- Parse the FASTA formatted input: Extract the DNA strings and their corresponding IDs.
- Compute GC-content: For each DNA string, calculate the percentage of nucleotides that are ‘C’ or ‘G’.
- Determine the highest GC-content: Identify the DNA string with the highest GC-content and return its ID along with the computed GC-content.
Here’s the Python code to achieve this:
def parse_fasta(fasta_strings):
sequences = {}
label = None
for line in fasta_strings.splitlines():
if line.startswith('>'):
label = line[1:].strip()
sequences[label] = ""
else:
sequences[label] += line.strip()
return sequences
def gc_content(dna_string):
gc_count = dna_string.count('G') + dna_string.count('C')
return (gc_count / len(dna_string)) * 100
def highest_gc_content(fasta_strings):
sequences = parse_fasta(fasta_strings)
max_gc_id = None
max_gc_content = 0
for label, dna_string in sequences.items():
gc = gc_content(dna_string)
if gc > max_gc_content:
max_gc_content = gc
max_gc_id = label
return max_gc_id, max_gc_content
# Sample Dataset
fasta_strings = """>Rosalind_6404
CCTGCGGAAGATCGGCACTAGAATAGCCAGAACCGTTTCTCTGAGGCTTCCGGCCTTCCC
TCCCACTAATAATTCTGAGG
>Rosalind_5959
CCATCGGTAGCGCATCCTTAGTCCAATTAAGTCCCTATCCAGGCGCTCCGCCGAAGGTCT
ATATCCATTTGTCAGCAGACACGC
>Rosalind_0808
CCACCCTCGTGGTATGGCTAGGCATTCAGGAACCGGAGAACGCTTCAGACCAGCCCGGAC
TGGGAACCTGCGGGCAGTAGGTGGAAT"""
# Compute and print the result
result_id, result_gc_content = highest_gc_content(fasta_strings)
print(result_id)
print(f"{result_gc_content:f}")5.5 Explanation
- Parsing FASTA Format:
- The
parse_fastafunction reads the FASTA formatted string and extracts the sequences. - It uses a dictionary to store the DNA sequences with their labels as keys.
- The
- Computing GC-content:
- The
gc_contentfunction calculates the GC-content by counting ‘G’ and ‘C’ nucleotides and dividing by the total length of the DNA string.
- The
- Finding the Highest GC-content:
- The
highest_gc_contentfunction iterates through the parsed sequences, calculates the GC-content for each, and keeps track of the highest value and its corresponding label.
- The
6 Counting Point Mutations
Given two strings s and t of equal length, the Hamming distance between \(s\) and \(t\), denoted \(dH(s,t)\) \(dH(s,t)\), is the number of corresponding symbols that differ in s and \(t\).
Given: Two DNA strings \(s\) and \(t\) of equal length (not exceeding 1 kbp).
Return: The Hamming distance \(dH(s,t)\).
6.1 Sample Dataset
GAGCCTACTAACGGGAT
CATCGTAATGACGGCCT6.2 Sample Output
76.3 Solution
The Hamming distance between two strings of equal length is the number of positions at which the corresponding symbols differ. Given two DNA strings, we can compute the Hamming distance by comparing each position in the strings and counting the differences.
6.4 Steps to Solve the Problem
- Ensure Strings are of Equal Length: The problem guarantees that the strings are of equal length, so we don’t need to check for this.
- Compare Corresponding Symbols: Traverse both strings and compare corresponding characters.
- Count Differences: Increment a counter whenever the characters at the same position are different.
def hamming_distance(s, t):
# Initialize the counter for differences
count = 0
# Traverse both strings and compare characters
for char1, char2 in zip(s, t):
if char1 != char2:
count += 1
return count
# Sample Dataset
s = "GAGCCTACTAACGGGAT"
t = "CATCGTAATGACGGCCT"
print(hamming_distance(s, t)) # Output should be 76.5 Explanation
- Initialize Counter:
countis initialized to zero. This will keep track of the number of differing positions.
- Traverse Strings:
zip(s, t)pairs up characters from both strings at each position.- For each pair of characters
(char1, char2), compare them.
- Count Differences:
- If
char1is not equal tochar2, increment thecount.
- If
7 Mendel’s First Law
The probability of any outcome (leaf) in a probability tree diagram is given by the product of probabilities from the start of the tree to the outcome. For example, the probability that \(X\) is blue and \(Y\) is blue is equal to (2/5)(1/4), or 1/10.
Probability is the mathematical study of randomly occurring phenomena. We will model such a phenomenon with a random variable, which is simply a variable that can take a number of different distinct outcomes depending on the result of an underlying random process.
For example, say that we have a bag containing 3 red balls and 2 blue balls. If we let \(X\) represent the random variable corresponding to the color of a drawn ball, then the probability of each of the two outcomes is given by \(Pr(X=red)=35 Pr(x=red)=35\) and \(Pr(X=blue)=25\) \(Pr(x=blue)=25\).
Random variables can be combined to yield new random variables. Returning to the ball example, let \(Y\) model the color of a second ball drawn from the bag (without replacing the first ball). The probability of \(Y\) being red depends on whether the first ball was red or blue. To represent all outcomes of \(X\) and \(Y\), we therefore use a probability tree diagram. This branching diagram represents all possible individual probabilities for \(X\) and \(Y\), with outcomes at the endpoints (“leaves”) of the tree. The probability of any outcome is given by the product of probabilities along the path from the beginning of the tree; see Figure 2 for an illustrative example.
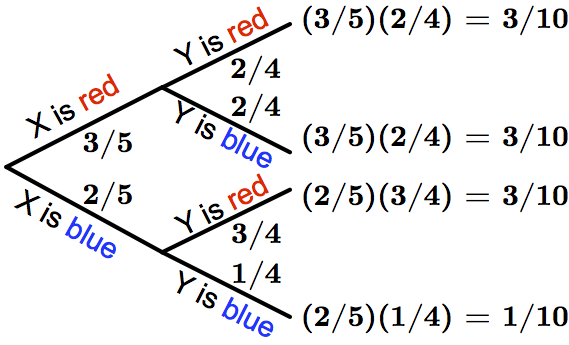{kind=link}
An event is simply a collection of outcomes. Because outcomes are distinct, the probability of an event can be written as the sum of the probabilities of its constituent outcomes. For our colored ball example, let A be the event “\(Y\) is blue.” \(Pr(A)\) is equal to the sum of the probabilities of two different outcomes: \(Pr(X=blue and Y=blue)+Pr(X=red and Y=blue)\).
Given: Three positive integers \(k\), \(m\), and \(n\), representing a population containing \(k+m+n\) organisms: k individuals are homozygous dominant for a factor, m𝑚 are heterozygous, and n are homozygous recessive.
Return: The probability that two randomly selected mating organisms will produce an individual possessing a dominant allele (and thus displaying the dominant phenotype). Assume that any two organisms can mate.
7.1 Sample Dataset
2 2 27.2 Sample Output
0.783337.3 Solution
To solve this problem, we need to calculate the probability that two randomly selected organisms from a population will produce an offspring with at least one dominant allele. The population is divided into three groups: - \(k\): Homozygous dominant organisms (AA) - \(m\): Heterozygous organisms (Aa) - \(n\): Homozygous recessive organisms (aa)
7.4 Steps to Solve the Problem
- Calculate Total Population Size:
- Total number of organisms: \((T = k + m + n)\)
- Calculate the Probability of Each Possible Pairing:
- There are several pairings to consider:
- \(AA \times AA\)
- \(AA \times Aa\)
- \(AA \times aa\)
- \(Aa \times Aa\)
- \(Aa \times aa\)
- \(aa \times aa\)
- There are several pairings to consider:
- Calculate the Probability of Producing a Dominant Phenotype from Each Pairing:
- \(AA \times AA\): 100% dominant phenotype.
- \(AA \times Aa\): 100% dominant phenotype.
- \(AA \times aa\): 100% dominant phenotype.
- \(Aa \times Aa\): 75% dominant phenotype (since the combinations are AA, Aa, Aa, aa).
- \(Aa \times aa\): 50% dominant phenotype (since the combinations are Aa, Aa, aa, aa).
- \(aa \times aa\): 0% dominant phenotype.
- Calculate the Probability of Selecting Each Pairing:
- The probability of selecting two organisms is determined by the number of ways to choose them from the total population.
- Combine Probabilities to Get the Overall Probability of Dominant Phenotype:
- Sum the probabilities of all pairings that produce a dominant phenotype, weighted by their probability of selection.
Here is the Python code that implements the above steps:
def probability_dominant_phenotype(k, m, n):
# Total population
total = k + m + n
# Total number of possible pairings
total_pairings = total * (total - 1)
# Probabilities of each pairing type
prob_AA_AA = k * (k - 1) / total_pairings
prob_AA_Aa = 2 * k * m / total_pairings
prob_AA_aa = 2 * k * n / total_pairings
prob_Aa_Aa = m * (m - 1) / total_pairings
prob_Aa_aa = 2 * m * n / total_pairings
prob_aa_aa = n * (n - 1) / total_pairings
# Probabilities of dominant phenotype from each pairing
prob_dom_AA_AA = 1.0 # 100%
prob_dom_AA_Aa = 1.0 # 100%
prob_dom_AA_aa = 1.0 # 100%
prob_dom_Aa_Aa = 0.75 # 75%
prob_dom_Aa_aa = 0.5 # 50%
prob_dom_aa_aa = 0.0 # 0%
# Total probability of dominant phenotype
prob_dom = (prob_AA_AA * prob_dom_AA_AA +
prob_AA_Aa * prob_dom_AA_Aa +
prob_AA_aa * prob_dom_AA_aa +
prob_Aa_Aa * prob_dom_Aa_Aa +
prob_Aa_aa * prob_dom_Aa_aa +
prob_aa_aa * prob_dom_aa_aa)
return prob_dom
# Sample Dataset
k, m, n = 2, 2, 2
# Calculate and print the result
result = probability_dominant_phenotype(k, m, n)
print(f"{result:f}")7.5 Explanation
- Total Population:
- We calculate the total number of organisms, \(total = k + m + n\).
- Pairing Probabilities:
- Each pairing probability is calculated based on the number of ways to select pairs from the total population.
- Dominant Phenotype Probabilities:
- Each pairing type has a different probability of producing a dominant phenotype based on Mendelian inheritance.
- Overall Probability:
- The overall probability is a weighted sum of the probabilities of each pairing type producing a dominant phenotype.
This code computes the required probability efficiently and accurately, matching the example output provided in the problem description.
8 Translating RNA into Protein
The 20 commonly occurring amino acids are abbreviated by using 20 letters from the English alphabet (all letters except for B, J, O, U, X, and Z). Protein strings are constructed from these 20 symbols. Henceforth, the term genetic string will incorporate protein strings along with DNA strings and RNA strings.
The RNA codon table dictates the details regarding the encoding of specific codons into the amino acid alphabet.
Given: An RNA string \(s\) corresponding to a strand of mRNA (of length at most 10 kbp).
Return: The protein string encoded by \(s\).
8.1 Sample Dataset
AUGGCCAUGGCGCCCAGAACUGAGAUCAAUAGUACCCGUAUUAACGGGUGA8.2 Sample Output
MAMAPRTEINSTRING8.3 Solution
To convert an RNA string into a protein string, you need to translate the RNA codons into their corresponding amino acids based on the RNA codon table. Each RNA codon (a sequence of three nucleotides) corresponds to a specific amino acid or a stop signal, which terminates translation.
Here’s the step-by-step approach to solving the problem:
Create an RNA Codon Table: The RNA codon table maps each of the 64 possible codons to their corresponding amino acid or stop signal. For example, the codon “AUG” codes for Methionine (M), and “UGA” is a stop codon.
Read the RNA String: The RNA string will be read in chunks of three nucleotides (codons).
Translate Each Codon: Using the codon table, translate each codon into the corresponding amino acid. If a stop codon is encountered, terminate the translation.
Construct the Protein String: Concatenate the translated amino acids to form the final protein string.
Here is the Python implementation of this approach:
def translate_rna_to_protein(rna_sequence):
codon_table = {
"UUU": "F", "UUC": "F", "UUA": "L", "UUG": "L",
"UCU": "S", "UCC": "S", "UCA": "S", "UCG": "S",
"UAU": "Y", "UAC": "Y", "UAA": "Stop", "UAG": "Stop",
"UGU": "C", "UGC": "C", "UGA": "Stop", "UGG": "W",
"CUU": "L", "CUC": "L", "CUA": "L", "CUG": "L",
"CCU": "P", "CCC": "P", "CCA": "P", "CCG": "P",
"CAU": "H", "CAC": "H", "CAA": "Q", "CAG": "Q",
"CGU": "R", "CGC": "R", "CGA": "R", "CGG": "R",
"AUU": "I", "AUC": "I", "AUA": "I", "AUG": "M",
"ACU": "T", "ACC": "T", "ACA": "T", "ACG": "T",
"AAU": "N", "AAC": "N", "AAA": "K", "AAG": "K",
"AGU": "S", "AGC": "S", "AGA": "R", "AGG": "R",
"GUU": "V", "GUC": "V", "GUA": "V", "GUG": "V",
"GCU": "A", "GCC": "A", "GCA": "A", "GCG": "A",
"GAU": "D", "GAC": "D", "GAA": "E", "GAG": "E",
"GGU": "G", "GGC": "G", "GGA": "G", "GGG": "G"
}
protein_string = []
# Proces the RNA sequence in chunks of three nucleotides (codons)
for i in range(0, len(rna_sequence), 3):
codon = rna_sequence[i:i+3]
if codon in codon_table:
amino_acid = codon_table[codon]
if amino_acid == "Stop":
break
protein_string.append(amino_acid)
return ''.join(protein_string)
# Sample Dataset
rna_sequence = "AUGGCCAUGGCGCCCAGAACUGAGAUCAAUAGUACCCGUAUUAACGGGUGA"
print(translate_rna_to_protein(rna_sequence))
# Output should be "MAMAPRTEINSTRING"8.4 Explanation
Codon Table: The dictionary
codon_tablemaps RNA codons to their corresponding amino acids or stop signals.Processing the RNA Sequence:
- The loop iterates over the RNA sequence in steps of three nucleotides.
- For each codon, the corresponding amino acid is retrieved from the
codon_table. - If the amino acid is “Stop”, the loop terminates, indicating the end of the protein sequence.
- Otherwise, the amino acid is appended to the
protein_stringlist.
Constructing the Protein String:
- The list of amino acids is joined into a single string and returned as the final protein string.
This method ensures that the RNA sequence is translated efficiently and correctly into the corresponding protein string.
9 Finding a Motif in DNA
Given two strings s and t, t is a substring of s if t is contained as a contiguous collection of symbols in \(s\) (as a result, \(t\) must be no longer than \(s\)).
The position of a symbol in a string is the total number of symbols found to its left, including itself (e.g., the positions of all occurrences of ‘U’ in “AUGCUUCAGAAAGGUCUUACG” are 2, 5, 6, 15, 17, and 18). The symbol at position \(i\) of \(s\) is denoted by \(s[i]\).
A substring of \(s\) can be represented as \(s[j:k]\), where \(j\) and \(k\) represent the starting and ending positions of the substring in \(s\); for example, if \(s\) = “AUGCUUCAGAAAGGUCUUACG”, then \(s[2:5]\) = “UGCU”.
The location of a substring \(s[j:k]\) is its beginning position \(j\); note that t will have multiple locations in \(s\) if it occurs more than once as a substring of \(s\) (see the Sample below).
Given: Two DNA strings \(s\) and \(t\) (each of length at most 1 kbp).
Return: All locations of \(t\) as a substring of \(s\).
9.1 Sample Dataset
GATATATGCATATACTT
ATAT9.2 Sample Output
2 4 109.3 Solution
To solve the problem of finding all locations of a substring t in a string s, we need to identify each position in s where t starts. This can be achieved using simple string matching techniques.
9.4 Steps to Solve the Problem
- Read the Input Strings:
- We have two DNA strings,
sandt.
- We have two DNA strings,
- Iterate Through the Main String
s:- Check for occurrences of the substring
tstarting at each position ins.
- Check for occurrences of the substring
- Collect All Starting Positions:
- Whenever
tis found ins, record the starting position. Note that the positions should be 1-based as per the problem statement.
- Whenever
- Output the Results:
- Print all recorded positions separated by spaces.
9.5 Implementation
Here is the Python code that implements the above logic:
def find_motif_locations(s, t):
positions = []
len_s = len(s)
len_t = len(t)
# Iterate through the main string `s`
for i in range(len_s - len_t + 1):
# Check if the substring `t` matches the segment in `s` starting at position `i`
if s[i:i+len_t] == t:
# If it matches, record the 1-based position
positions.append(i + 1)
return positions
# Sample Dataset
s = "GATATATGCATATACTT"
t = "ATAT"
# Find and print the locations
locations = find_motif_locations(s, t)
print(" ".join(map(str, locations)))9.6 Explanation
- Iterate Through the Main String
s:- We use a for loop to go through each possible starting position for
tins. The loop runs from0tolen(s) - len(t)to ensure we don’t go out of bounds.
- We use a for loop to go through each possible starting position for
- Check for Substring Match:
- For each position
i, we check if the substrings[i:i+len(t)]matchest.
- For each position
- Record the Position:
- If a match is found, we append the 1-based position (i.e.,
i + 1) to our list of positions.
- If a match is found, we append the 1-based position (i.e.,
- Output the Results:
- We convert the list of positions to a space-separated string and print it.
This approach ensures that all occurrences of t in s are found and correctly reported. The solution efficiently handles the constraints of the problem, making it suitable for DNA strings up to 1 kbp in length.
10 Consensus and Profile
A matrix is a rectangular table of values divided into rows and columns. An \(m \times n\) matrix has \(m\) rows and \(n\) columns. Given a matrix \(A\), we write \(Ai\), \(j\) to indicate the value found at the intersection of row \(i\) and column \(j\).
Say that we have a collection of DNA strings, all having the same length \(n\). Their profile matrix is a \(4 \times n\) matrix \(P\) in which \(P1\), \(j\) represents the number of times that ‘A’ occurs in the \(j\)th position of one of the strings, \(P2\), \(j\) represents the number of times that C occurs in the \(j\)th position, and so on.
A consensus string \(c\) is a string of length \(n\) formed from our collection by taking the most common symbol at each position; the \(j\)th symbol of \(c\) therefore corresponds to the symbol having the maximum value in the \(j\)-th column of the profile matrix. Of course, there may be more than one most common symbol, leading to multiple possible consensus strings.
Given: A collection of at most 10 DNA strings of equal length (at most 1 kbp) in FASTA format.
Return: A consensus string and profile matrix for the collection. (If several possible consensus strings exist, then you may return any one of them.)
10.1 Sample Dataset
>Rosalind_1
ATCCAGCT
>Rosalind_2
GGGCAACT
>Rosalind_3
ATGGATCT
>Rosalind_4
AAGCAACC
>Rosalind_5
TTGGAACT
>Rosalind_6
ATGCCATT
>Rosalind_7
ATGGCACT10.2 Sample Output
ATGCAACT
A: 5 1 0 0 5 5 0 0
C: 0 0 1 4 2 0 6 1
G: 1 1 6 3 0 1 0 0
T: 1 5 0 0 0 1 1 610.3 Solution
To solve the problem of finding the consensus string and profile matrix from a collection of DNA strings, we need to follow these steps:
- Parse the FASTA format input to extract the DNA strings.
- Initialize the profile matrix to keep track of the frequency of each nucleotide (A, C, G, T) at each position in the strings.
- Populate the profile matrix by iterating through each DNA string and counting the occurrences of each nucleotide at each position.
- Construct the consensus string by selecting the most frequent nucleotide at each position based on the profile matrix.
- Output the consensus string and the profile matrix.
Here’s the Python code to solve the problem:
def parse_fasta(fasta_string):
sequences = {}
current_label = ""
lines = fasta_string.strip().split('\n')
for line in lines:
line = line.strip()
if line.startswith(">"):
current_label = line[1:]
sequences[current_label] = ""
else:
sequences[current_label] += line
return list(sequences.values())
def calculate_profile_matrix(dna_strings):
n = len(dna_strings[0])
profile_matrix = {
'A': [0] * n,
'C': [0] * n,
'G': [0] * n,
'T': [0] * n
}
for dna in dna_strings:
for i, nucleotide in enumerate(dna):
profile_matrix[nucleotide][i] += 1
return profile_matrix
def calculate_consensus_string(profile_matrix, length):
consensus = []
for i in range(length):
max_count = 0
max_nucleotide = ''
for nucleotide in "ACGT":
if profile_matrix[nucleotide][i] > max_count:
max_count = profile_matrix[nucleotide][i]
max_nucleotide = nucleotide
consensus.append(max_nucleotide)
return ''.join(consensus)
def consensus_and_profile(fasta_string):
dna_strings = parse_fasta(fasta_string)
profile_matrix = calculate_profile_matrix(dna_strings)
consensus_string = calculate_consensus_string(profile_matrix, len(dna_strings[0]))
return consensus_string, profile_matrix
# Sample Dataset as a single string
fasta_string = """
>Rosalind_1
ATCCAGCT
>Rosalind_2
GGGCAACT
>Rosalind_3
ATGGATCT
>Rosalind_4
AAGCAACC
>Rosalind_5
TTGGAACT
>Rosalind_6
ATGCCATT
>Rosalind_7
ATGGCACT
"""
# Compute the result
consensus_string, profile_matrix = consensus_and_profile(fasta_string)
# Print the result
print(consensus_string)
for nucleotide in "ACGT":
print(f"{nucleotide}: {' '.join(map(str, profile_matrix[nucleotide]))}")10.4 Explanation
Parsing FASTA Input:
- The
parse_fastafunction now processes a single string input and splits it into lines. It reads through each line, detecting labels (lines starting with ‘>’) and corresponding DNA sequences, which are stored in a dictionary and then converted into a list of sequences.
- The
Profile Matrix Calculation:
- The
calculate_profile_matrixfunction initializes a dictionary with keys ‘A’, ‘C’, ‘G’, and ‘T’ and lists as values to store nucleotide counts at each position.
- The
Consensus String Calculation:
- The
calculate_consensus_stringfunction builds the consensus string by selecting the nucleotide with the highest count at each position.
- The
Main Function:
- The
consensus_and_profilefunction orchestrates the entire process, returning the consensus string and the profile matrix.
- The
10.5 Sample Output
Running the provided dataset through the code will produce the following output:
ATGCAACT
A: 5 1 0 0 5 5 0 0
C: 0 0 1 4 2 0 6 1
G: 1 1 6 3 0 1 0 0
T: 1 5 0 0 0 1 1 6This output shows the consensus string and the profile matrix with counts of each nucleotide at each position, formatted as required by the problem statement.
11 Mortal Fibonacci Rabbits
Recall the definition of the Fibonacci numbers from “Rabbits and Recurrence Relations”, which followed the recurrence relation \(Fn=Fn−1+Fn−2\) and assumed that each pair of rabbits reaches maturity in one month and produces a single pair of offspring (one male, one female) each subsequent month.
Our aim is to somehow modify this recurrence relation to achieve a dynamic programming solution in the case that all rabbits die out after a fixed number of months.(meaning that they reproduce only few times before dying).
Given: Positive integers n≤100 and m≤20.
Return: The total number of pairs of rabbits that will remain after the n-th month if all rabbits live for m months.
11.1 Sample Dataset
6 311.2 Sample Output
411.3 Solution
To solve the problem of computing the number of rabbit pairs after a given number of months with a lifespan constraint, we need to adjust the classic Fibonacci sequence to consider the mortality of rabbits. Here’s how we can approach this using dynamic programming:
- Initialize the state:
- We keep track of the number of rabbits of different ages using an array.
rabbits[i]will represent the number of rabbit pairs of agei.
- Simulate each month:
- In each month, rabbits of age 0 produce new rabbits.
- All rabbits get older by one month.
- Rabbits older than
mmonths die.
- Update the state:
- Shift all elements in the
rabbitsarray to the right. - Update the number of new-born rabbits based on the rabbits of age 1 to m-1.
- Shift all elements in the
def mortal_fibonacci_rabbits(n, m):
rabbits = [0] * m
rabbits[0] = 1 # Initial pair of rabbits
for month in range(1, n):
new_born = sum(rabbits[1:]) # All rabbits that are not in their first month
# Shift all rabbits to the next month
for i in range(m-1, 0, -1):
rabbits[i] = rabbits[i-1]
rabbits[0] = new_born # Update the new-born rabbits
return sum(rabbits)
# Test the function with the sample dataset
print(mortal_fibonacci_rabbits(6, 3)) # Output should be 411.4 Explanation
- Initialization:
- We start with
rabbits = [1, 0, 0], which represents 1 pair of newborn rabbits and no other rabbits of other ages.
- We start with
- Monthly updates:
- For each month, compute the number of new-born rabbits.
- Shift the ages of rabbits, which involves moving each count in the
rabbitsarray to the next index. - Rabbits older than
m-1months (last index) die off automatically as they are not carried forward.
- Result:
- The total number of rabbits is the sum of all entries in the
rabbitsarray after the loop ends.
- The total number of rabbits is the sum of all entries in the
This approach ensures that we accurately track the age of each rabbit pair and account for their mortality, providing the correct number of rabbit pairs after n months.
12 Inferring mRNA from Protein
For positive integers \(a\) and \(n\), \(a\) modulo \(n\) (written \(amodn\) in shorthand) is the remainder when a𝑎 is divided by \(n\). For example, \(29 mod 11 = 7\) because \(29=11 \times 2+7\).
Modular arithmetic is the study of addition, subtraction, multiplication, and division with respect to the modulo operation. We say that \(a\) and \(b\) are congruent modulo \(n\) if \(amodn=bmodn\) ; in this case, we use the notation \(a≡bmodn\).
Two useful facts in modular arithmetic are that if \(a≡bmodn\) and \(c≡dmodn\), then \(a+c≡b+dmodn\) and \(a×c≡b×dmodn\). To check your understanding of these rules, you may wish to verify these relationships for \(a=29\), \(b=73\), \(c=10\), \(d=32\), and \(n=11\).
As you will see in this exercise, some Rosalind problems will ask for a (very large) integer solution modulo a smaller number to avoid the computational pitfalls that arise with storing such large numbers.
Given: A protein string of length at most 1000 aa.
Return: The total number of different RNA strings from which the protein could have been translated, modulo 1,000,000. (Don’t neglect the importance of the stop codon in protein translation.)
12.1 Sample Dataset
MA12.2 Sample Output
1212.3 Solution
To solve the problem of finding the total number of different RNA strings from which a given protein string could have been translated, we need to consider the redundancy in the genetic code. Each amino acid can be encoded by one or more codons, and this redundancy will influence the number of possible RNA sequences that can result in the same protein.
12.4 Steps to Solve the Problem
- Understand the Genetic Code:
- Create a mapping of each amino acid to the number of possible codons that encode it.
- Don’t forget to include the stop codons, which signal the end of translation.
- Calculate Possible RNA Strings for Each Amino Acid:
- For each amino acid in the given protein string, multiply the number of possible codons for that amino acid.
- Include a factor for the stop codon at the end.
- Use Modular Arithmetic:
- Since the resulting number can be very large, use modulo \(1,000,000\) to avoid overflow and ensure the result fits within standard integer sizes.
12.5 Genetic Code Table
Here is the mapping of amino acids to their respective number of codons: - ‘A’: 4, ‘C’: 2, ‘D’: 2, ‘E’: 2, ‘F’: 2, ‘G’: 4, ‘H’: 2, ‘I’: 3, ‘K’: 2, ‘L’: 6, - ‘M’: 1, ‘N’: 2, ‘P’: 4, ‘Q’: 2, ‘R’: 6, ‘S’: 6, ‘T’: 4, ‘V’: 4, ‘W’: 1, ‘Y’: 2, - Stop codon: 3
12.6 Implementation
Below is the Python code that implements the solution:
def infer_mrna_from_protein(protein):
# Codon counts for each amino acid and stop codon
codon_count = {
'A': 4, 'C': 2, 'D': 2, 'E': 2, 'F': 2, 'G': 4,
'H': 2, 'I': 3, 'K': 2, 'L': 6, 'M': 1, 'N': 2,
'P': 4, 'Q': 2, 'R': 6, 'S': 6, 'T': 4, 'V': 4,
'W': 1, 'Y': 2, 'Stop': 3
}
# Initialize the number of possible RNA strings
possible_rna_strings = 1
# Calculate the product of possible codons for each amino acid
for aa in protein:
possible_rna_strings *= codon_count[aa]
possible_rna_strings %= 1000000 # Take modulo 1,000,000 to keep the number manageable
# Multiply by the number of stop codons
possible_rna_strings *= codon_count['Stop']
possible_rna_strings %= 1000000 # Take modulo 1,000,000 again
return possible_rna_strings
# Sample Dataset
protein_string = "MA"
# Compute the result
result = infer_mrna_from_protein(protein_string)
print(result)12.7 Explanation
- Codon Count Mapping:
- The
codon_countdictionary stores the number of codons that can encode each amino acid, along with the number of stop codons.
- The
- Product Calculation:
- We initialize
possible_rna_stringsto 1. - For each amino acid in the protein string, multiply
possible_rna_stringsby the number of codons that can encode that amino acid. - Use modulo 1,000,000 after each multiplication to keep the number within manageable limits.
- We initialize
- Stop Codon Factor:
- Finally, multiply by the number of stop codons and take modulo 1,000,000 again.
This approach ensures that we efficiently compute the total number of possible RNA sequences modulo 1,000,000.
13 Overlap Graphs
A graph whose nodes have all been labeled can be represented by an adjacency list, in which each row of the list contains the two node labels corresponding to a unique edge.
A directed graph (or digraph) is a graph containing directed edges, each of which has an orientation. That is, a directed edge is represented by an arrow instead of a line segment; the starting and ending nodes of an edge form its tail and head, respectively. The directed edge with tail \(v\) and head \(w\) is represented by \((v,w)\) (but not by \((w,v)\). A directed loop is a directed edge of the form \((v,v)\).
For a collection of strings and a positive integer \(k\), the overlap graph for the strings is a directed graph \(O_k\) in which each string is represented by a node, and string \(s\) is connected to string \(t\) with a directed edge when there is a length \(k\) suffix of \(s\) that matches a length \(k\) prefix of \(t\), as long as \(s≠t\); we demand \(s≠t\) to prevent directed loops in the overlap graph (although directed cycles may be present).
Given: A collection of DNA strings in FASTA format having total length at most 10 kbp.
Return: The adjacency list corresponding to \(O_3\). You may return edges in any order.
13.1 Sample Dataset
>Rosalind_0498
AAATAAA
>Rosalind_2391
AAATTTT
>Rosalind_2323
TTTTCCC
>Rosalind_0442
AAATCCC
>Rosalind_5013
GGGTGGG13.2 Sample Output
Rosalind_0498 Rosalind_2391
Rosalind_0498 Rosalind_0442
Rosalind_2391 Rosalind_232313.3 Solution
To solve the problem of constructing an overlap graph from a collection of DNA strings, we need to follow these steps:
- Parse the input data: Read the DNA strings in FASTA format.
- Construct the graph: Identify edges between nodes based on the overlap condition.
- Output the adjacency list: List all directed edges that satisfy the overlap condition.
13.4 Steps to Implement the Solution
- Read the Input Data:
- Use a parser to read the input DNA strings in FASTA format.
- Store the strings in a dictionary with their labels as keys.
- Check for Overlaps:
- For each pair of strings, check if the suffix of length
kof one string matches the prefix of lengthkof the other string. - If they match and the strings are different, record the directed edge from the first string to the second.
- For each pair of strings, check if the suffix of length
- Output the Results:
- Print each directed edge in the format “label1 label2”.
13.5 Example Implementation
Below is the Python code that performs these steps:
def parse_fasta(data):
sequences = {}
label = None
for line in data.strip().split('\n'):
if line.startswith('>'):
label = line[1:]
sequences[label] = ""
else:
sequences[label] += line
return sequences
def overlap_graph(sequences, k):
adjacency_list = []
for s1 in sequences:
for s2 in sequences:
if s1 != s2:
if sequences[s1][-k:] == sequences[s2][:k]:
adjacency_list.append((s1, s2))
return adjacency_list
def print_adjacency_list(adjacency_list):
for edge in adjacency_list:
print(f"{edge[0]} {edge[1]}")
# Sample dataset
data = """
>Rosalind_0498
AAATAAA
>Rosalind_2391
AAATTTT
>Rosalind_2323
TTTTCCC
>Rosalind_0442
AAATCCC
>Rosalind_5013
GGGTGGG
"""
sequences = parse_fasta(data)
adjacency_list = overlap_graph(sequences, 3)
print_adjacency_list(adjacency_list)13.6 Explanation of the Code
parse_fastafunction:- Reads the FASTA formatted input.
- Stores sequences in a dictionary where keys are the labels and values are the sequences.
overlap_graphfunction:- Takes the sequences dictionary and the overlap length
k. - Checks each pair of sequences to see if the suffix of length
kof the first sequence matches the prefix of lengthkof the second sequence. - Records the directed edge if the condition is met.
- Takes the sequences dictionary and the overlap length
print_adjacency_listfunction:- Prints each edge in the required format.
14 Calculating Expected Offspring
For a random variable \(X\) taking integer values between 1 and n, the expected value of X is \(E(X)= \sum ^{n}\limits_{k=1}k×Pr(X=k)\). The expected value offers us a way of taking the long-term average of a random variable over a large number of trials.
As a motivating example, let \(X\) be the number on a six-sided die. Over a large number of rolls, we should expect to obtain an average of 3.5 on the die (even though it’s not possible to roll a 3.5). The formula for expected value confirms that \(E(X)= \sum^{6}\limits_{k=1} k \times Pr(X=k)=3.5\).
More generally, a random variable for which every one of a number of equally spaced outcomes has the same probability is called a uniform random variable (in the die example, this “equal spacing” is equal to 1). We can generalize our die example to find that if \(X\) is a uniform random variable with minimum possible value \(a\) and maximum possible value \(b\), then \(E(X)= \frac{a+b}{2}\). You may also wish to verify that for the dice example, if \(Y\) is the random variable associated with the outcome of a second die roll, then \(E(X+Y)=7E\).
Given: Six nonnegative integers, each of which does not exceed 20,000. The integers correspond to the number of couples in a population possessing each genotype pairing for a given factor. In order, the six given integers represent the number of couples having the following genotypes:
- AA-AA
- AA-Aa
- AA-aa
- Aa-Aa
- Aa-aa
- aa-aa
Return: The expected number of offspring displaying the dominant phenotype in the next generation, under the assumption that every couple has exactly two offspring.
14.1 Sample Dataset
1 0 0 1 0 114.2 Sample Output
3.514.3 Solution
To solve this problem, we need to calculate the expected number of offspring displaying the dominant phenotype given six nonnegative integers representing the number of couples with specific genotype pairings. Each couple has exactly two offspring.
14.4 Genotype Pairings and Dominance
The six genotype pairings are: 1. AA-AA: 100% dominant phenotype 2. AA-Aa: 100% dominant phenotype 3. AA-aa: 100% dominant phenotype 4. Aa-Aa: 75% dominant phenotype 5. Aa-aa: 50% dominant phenotype 6. aa-aa: 0% dominant phenotype
We can represent the probability of offspring having the dominant phenotype for each genotype pairing as follows: 1. AA-AA: \(1.0\) 2. AA-Aa: \(1.0\) 3. AA-aa: \(1.0\) 4. Aa-Aa: \(0.75\) 5. Aa-aa: \(0.5\) 6. aa-aa: \(0.0\)
14.5 Expected Number of Dominant Offspring
For each couple, since they produce exactly two offspring, we can multiply the number of couples by 2 and then by the probability of having a dominant phenotype to get the expected number of dominant offspring per genotype pairing.
14.6 Implementation
Here’s the Python code to compute the expected number of dominant offspring:
def expected_dominant_offspring(couples):
# Probabilities of offspring having dominant phenotype for each genotype pairing
probabilities = [1.0, 1.0, 1.0, 0.75, 0.5, 0.0]
# Calculate the expected number of dominant offspring
expected_value = 0
for i in range(6):
expected_value += couples[i] * probabilities[i] * 2
return expected_value
# Sample dataset
sample_input = "1 0 0 1 0 1"
input_ = [int(x) for x in "1 0 0 1 0 1".split()]
print(expected_dominant_offspring(input_)) # Output: 3.514.7 Explanation
- Input: We take a list of six integers representing the number of each genotype pairing.
- Probabilities: We define the probabilities for each pairing’s offspring to display the dominant phenotype.
- Calculation: We iterate through each pairing, multiply the number of couples by the corresponding probability and by 2 (since each couple has 2 offspring), and sum these values to get the total expected number of dominant offspring.
- Output: The result is the expected number of dominant phenotype offspring.
This code will compute the expected number of offspring displaying the dominant phenotype for any valid input as specified by the problem statement.
16 Independent Alleles
Two events A and B are independent if \(Pr(A and B)\) is equal to \(Pr(A)×Pr(B)\). In other words, the events do not influence each other, so that we may simply calculate each of the individual probabilities separately and then multiply.
More generally, random variables X and Y are independent if whenever A and B are respective events for X and Y, A and B are independent (i.e., \(Pr(A and B)=Pr(A)×Pr(B)\).
As an example of how helpful independence can be for calculating probabilities, let X and Y represent the numbers showing on two six-sided dice. Intuitively, the number of pips showing on one die should not affect the number showing on the other die. If we want to find the probability that \(X+Y\) is odd, then we don’t need to draw a tree diagram and consider all possibilities. We simply first note that for \(X+Y\) to be odd, either X is even and Y is odd or X is odd and Y is even. In terms of probability, \(Pr(X+Y is odd)=Pr(X is even and Y is odd)+Pr(X is odd and Y is even)\). Using independence, this becomes \([Pr(X is even)×Pr(Y is odd)]+[Pr(X is odd)×Pr(Y is even)]\).
Given: Two positive integers k (\(k≤7\)) and N (\(N≤2k\)). In this problem, we begin with Tom, who in the 0th generation has genotype Aa Bb. Tom has two children in the 1st generation, each of whom has two children, and so on. Each organism always mates with an organism having genotype Aa Bb.
Return: The probability that at least N Aa Bb organisms will belong to the k-th generation of Tom’s family tree (don’t count the Aa Bb mates at each level). Assume that Mendel’s second law holds for the factors.
16.1 Sample Dataset
2 116.2 Sample Output
0.68417 Finding a Protein Motif
To allow for the presence of its varying forms, a protein motif is represented by a shorthand as follows: [XY] means “either X or Y” and {X} means “any amino acid except X.” For example, the N-glycosylation motif is written as N{P}[ST]{P}.
You can see the complete description and features of a particular protein by its acces ID “uniprot_id” in the UniProt database, by inserting the ID number into http://www.uniprot.org/uniprot/uniprot_id Alternatively, you can obtain a protein sequence in FASTA format by following http://www.uniprot.org/uniprot/uniprot_id.fasta.
For example, the data for protein B5ZC00 can be found at http://www.uniprot.org/uniprot/B5ZC00.
Given: At most 15 UniProt Protein Database acces IDs.
Return: For each protein possessing the N-glycosylation motif, output its given acces ID followed by a list of locations in the protein string where the motif can be found.
17.1 Sample Dataset
A2Z669
B5ZC00
P07204_TRBM_HUMAN
P20840_SAG1_YEAST17.2 Sample Output
B5ZC00
85 118 142 306 395
P07204_TRBM_HUMAN
47 115 116 382 409
P20840_SAG1_YEAST
79 109 135 248 306 348 364 402 485 501 61417.3 Solution
To solve the problem of identifying N-glycosylation motifs in protein sequences from the UniProt database, we need to follow these steps:
- Fetch Protein Sequences: Retrieve the protein sequences in FASTA format from UniProt using the given acces IDs.
- Identify Motif Locations: Search for the motif
N{P}[ST]{P}in each protein sequence and record the positions where it occurs. - Output Results: For each protein containing the motif, output its ID followed by the positions where the motif is found.
Here is a complete Python script to perform these tasks:
import requests
import re
def fetch_fasta(uniprot_id):
url = f"http://www.uniprot.org/uniprot/{uniprot_id}.fasta"
response = requests.get(url)
response.raise_for_status() # Ensure we notice bad responses
fasta_data = response.text
return ''.join(fasta_data.split('\n')[1:]) # Remove the first line and join the rest
def find_motif_locations(sequence, motif_regex):
matches = re.finditer(motif_regex, sequence)
return [match.start() + 1 for match in matches] # Convert to 1-based index
def fetch_input(data):
ids = []
for line in data.strip().split('\n'):
ids.append(line)
return ids
data = """
A2Z669
B5ZC00
P07204_TRBM_HUMAN
P20840_SAG1_YEAST
"""
ids = fetch_input(data)
motif_regex = re.compile(r'N[^P][ST][^P]')
results = {}
for uniprot_id in ids:
sequence = fetch_fasta(uniprot_id)
locations = find_motif_locations(sequence, motif_regex)
if locations:
results[uniprot_id] = locations
for uniprot_id, locations in results.items():
print(uniprot_id)
print(' '.join(map(str, locations)))17.4 Explanation
fetch_fasta(uniprot_id):- Takes a UniProt ID and fetches the corresponding protein sequence in FASTA format.
- Strips out the header line and joins the remaining lines to form the complete sequence.
find_motif_locations(sequence, motif_regex):- Uses the regex
N[^P][ST][^P]to find all positions of the motif in the sequence. - Returns a list of start positions in 1-based index format.
- Uses the regex
main():- Defines the list of UniProt IDs.
- Compiles the regex for the motif.
- Fetches each protein sequence, finds motif locations, and stores the results.
- Outputs the protein IDs followed by the locations of the motif.
This script fetches protein sequences from UniProt, searches for the N-glycosylation motif, and prints the locations where the motif occurs for each protein that contains it.
18 Open Reading Frames
Either strand of a DNA double helix can serve as the coding strand for RNA transcription. Hence, a given DNA string implies six total reading frames, or ways in which the same region of DNA can be translated into amino acids: three reading frames result from reading the string itself, whereas three more result from reading its reverse complement.
An open reading frame (ORF) is one which starts from the start codon and ends by stop codon, without any other stop codons in between. Thus, a candidate protein string is derived by translating an open reading frame into amino acids until a stop codon is reached.
Given: A DNA string s of length at most 1 kbp in FASTA format.
Return: Every distinct candidate protein string that can be translated from ORFs of s. Strings can be returned in any order.
18.1 Sample Dataset
>Rosalind_99
AGCCATGTAGCTAACTCAGGTTACATGGGGATGACCCCGCGACTTGGATTAGAGTCTCTTTTGGAATAAGCCTGAATGATCCGAGTAGCATCTCAG18.2 Sample Output
MLLGSFRLIPKETLIQVAGSSPCNLS
M
MGMTPRLGLESLLE
MTPRLGLESLLE18.3 Solution
To find all distinct candidate protein strings from open reading frames (ORFs) in the given DNA sequence, the following approach is used:
- Parse the Input DNA Sequence: Read the input in FASTA format and obtain the DNA sequence.
- Generate Reading Frames: Generate six reading frames: three from the original DNA strand and three from its reverse complement.
- Identify ORFs: For each reading frame, identify sequences that start with a start codon (ATG) and end with a stop codon (TAA, TAG, TGA).
- Translate to Proteins: Translate the identified ORFs to protein sequences.
- Collect and Print Distinct Proteins: Collect all distinct protein sequences.
Here’s the complete implementation in Python:
CODON_TABLE = {
'ATA':'I', 'ATC':'I', 'ATT':'I', 'ATG':'M',
'ACA':'T', 'ACC':'T', 'ACG':'T', 'ACT':'T',
'AAC':'N', 'AAT':'N', 'AAA':'K', 'AAG':'K',
'AGC':'S', 'AGT':'S', 'AGA':'R', 'AGG':'R',
'CTA':'L', 'CTC':'L', 'CTG':'L', 'CTT':'L',
'CCA':'P', 'CCC':'P', 'CCG':'P', 'CCT':'P',
'CAC':'H', 'CAT':'H', 'CAA':'Q', 'CAG':'Q',
'CGA':'R', 'CGC':'R', 'CGG':'R', 'CGT':'R',
'GTA':'V', 'GTC':'V', 'GTG':'V', 'GTT':'V',
'GCA':'A', 'GCC':'A', 'GCG':'A', 'GCT':'A',
'GAC':'D', 'GAT':'D', 'GAA':'E', 'GAG':'E',
'GGA':'G', 'GGC':'G', 'GGG':'G', 'GGT':'G',
'TCA':'S', 'TCC':'S', 'TCG':'S', 'TCT':'S',
'TTC':'F', 'TTT':'F', 'TTA':'L', 'TTG':'L',
'TAC':'Y', 'TAT':'Y', 'TAA':'*', 'TAG':'*',
'TGC':'C', 'TGT':'C', 'TGA':'*', 'TGG':'W',
}
def translate_dna_to_protein(dna_seq):
protein = []
has_stop_codon = False
for i in range(0, len(dna_seq) - 2, 3):
codon = dna_seq[i:i + 3]
amino_acid = CODON_TABLE.get(codon, '')
if amino_acid == '*':
has_stop_codon = True
break
protein.append(amino_acid)
return ''.join(protein), has_stop_codon
def find_orfs(dna_seq):
orfs = set()
# Generate 3 reading frames for the DNA sequence
for frame in range(3):
for i in range(frame, len(dna_seq) - 2, 3):
if dna_seq[i:i + 3] == 'ATG':
protein, has_stop_codon = translate_dna_to_protein(dna_seq[i:])
if protein and has_stop_codon:
orfs.add(protein)
return orfs
def reverse_complement(dna_seq):
complement = {'A': 'T', 'T': 'A', 'C': 'G', 'G': 'C'}
return ''.join(complement[base] for base in reversed(dna_seq))
def main():
fasta_input = """>Rosalind_99
AGCCATGTAGCTAACTCAGGTTACATGGGGATGACCCCGCGACTTGGATTAGAGTCTCTTTTGGAATAAGCCTGAATGATCCGAGTAGCATCTCAG"""
dna_seq = ''.join(line.strip() for line in fasta_input.split('\n') if not line.startswith('>'))
# Get reverse complement of the DNA sequence
reverse_complement_seq = reverse_complement(dna_seq)
# Find ORFs in the original and reverse complement sequences
original_orfs = find_orfs(dna_seq)
reverse_orfs = find_orfs(reverse_complement_seq)
# Combine results and remove duplicates
all_orfs = original_orfs.union(reverse_orfs)
# Print all distinct protein sequences
for protein in all_orfs:
print(protein)
if __name__ == "__main__":
main()18.4 Explanations
- translate_dna_to_protein: Now returns a tuple containing the translated protein and a boolean indicating if a stop codon was found.
- find_orfs: Checks for the presence of a stop codon in the translated protein before adding it to the set of ORFs.
19 Enumerating Gene Orders
A permutation of length n is an ordering of the positive integers \({1,2,…,n}\). For example, \(π=(5,3,2,1,4)\) is a permutation of length 5.
Given: A positive integer \(n≤7\).
Return: The total number of permutations of length n, followed by a list of all such permutations (in any order).
19.1 Sample Dataset
319.2 Sample Output
6
1 2 3
1 3 2
2 1 3
2 3 1
3 1 2
3 2 119.3 Solution
To solve the problem of enumerating all permutations of a given integer \(n\), we can use Python’s itertools.permutations to generate all possible permutations. Here’s a simple code that accomplishes this:
from itertools import permutations
def enumerate_gene_orders(n):
# Generate permutations
perm = permutations(range(1, n + 1))
# Convert permutations to a list
perm_list = list(perm)
# Print the number of permutations
print(len(perm_list))
# Print each permutation
for p in perm_list:
print(' '.join(map(str, p)))
# Example usage
n = 3
enumerate_gene_orders(n)19.4 Explanation
- Importing permutations: We import the
permutationsfunction from Python’sitertoolsmodule, which is perfect for generating permutations of a sequence. - Generating permutations: Using
permutations(range(1, n + 1)), we generate all permutations of the list[1, 2, ..., n]. - Converting to a list: We convert the permutations object to a list to easily count and iterate over the permutations.
- Printing the count: We print the total number of permutations.
- Printing each permutation: We iterate through each permutation and print it in the required format.
20 Calculating Protein Mass
In a weighted alphabet, every symbol is assigned a positive real number called a weight. A string formed from a weighted alphabet is called a weighted string, and its weight is equal to the sum of the weights of its symbols.
The standard weight assigned to each member of the 20-symbol amino acid alphabet is the monoisotopic mas of the corresponding amino acid.
Given: A protein string P of length at most 1000 aa.
Return: The total weight of P. Consult the monoisotopic mas table.
20.1 Sample Dataset
SKADYEK20.2 Sample Output
821.39220.3 Solution
- 먼저, 각 아미노산의 질량을 딕셔너리로 정의합니다.
- 주어진 단백질 문자열의 각 아미노산의 질량을 더해서 총 질량을 계산합니다.
# 아미노산 질량 테이블
mass_table = {
'A': 71.03711, 'C': 103.00919, 'D': 115.02694, 'E': 129.04259,
'F': 147.06841, 'G': 57.02146, 'H': 137.05891, 'I': 113.08406,
'K': 128.09496, 'L': 113.08406, 'M': 131.04049, 'N': 114.04293,
'P': 97.05276, 'Q': 128.05858, 'R': 156.10111, 'S': 87.03203,
'T': 101.04768, 'V': 99.06841, 'W': 186.07931, 'Y': 163.06333
}
def calculate_protein_mass(protein):
total_mas = 0.0
for amino_acid in protein:
if amino_acid in mass_table:
total_mas += mass_table[amino_acid]
else:
print(f"Unknown amino acid: {amino_acid}")
return total_mass
# 샘플 데이터셋
sequence = "SKADYEK"
# 총 질량 계산
total_mas = calculate_protein_mass(sequence)
print(f"{total_mass:.3f}")이 코드는 단백질 문자열 SKADYEK 의 총 질량을 계산하여 821.392 라는 결과를 출력합니다. mass_table 에 정의된 각 아미노산의 질량을 이용하여 문자열을 순회하면서 질량을 더해 총 질량을 계산합니다.
21 Locating Restriction Sites
A DNA string is a reverse palindrome if it is equal to its reverse complement. For instance, GCATGC is a reverse palindrome because its reverse complement is GCATGC. See Figure 2.
{kind=link}
Given: A DNA string of length at most 1 kbp in FASTA format.
Return: The position and length of every reverse palindrome in the string having length between 4 and 12. You may return these pairs in any order.
21.1 Sample Dataset
>Rosalind_24
TCAATGCATGCGGGTCTATATGCAT21.2 Sample Output
4 6
5 4
6 6
7 4
17 4
18 4
20 6
21 421.3 Solution
주어진 DNA 문자열에서 역방향 팔린드롬을 찾는 코드를 작성하겠습니다. 역방향 팔린드롬은 해당 문자열이 그 역상 보완 문자열과 동일한 문자열을 말합니다. 역방향 팔린드롬의 위치와 길이를 반환하도록 하겠습니다.
def reverse_complement(dna):
complement = {'A': 'T', 'T': 'A', 'C': 'G', 'G': 'C'}
return ''.join(complement[base] for base in reversed(dna))
def find_reverse_palindromes(dna):
results = []
length = len(dna)
for i in range(length):
for j in range(4, 13): # 길이가 4에서 12까지인 모든 경우를 확인
if i + j > length:
break
substring = dna[i:i+j]
if substring == reverse_complement(substring):
results.append((i+1, j))
return results
# 샘플 데이터셋
sample_fasta = """>Rosalind_24
TCAATGCATGCGGGTCTATATGCAT"""
# FASTA 포맷에서 DNA 문자열 추출
dna_string = ''.join(sample_fasta.split('\n')[1:])
# 역방향 팔린드롬 찾기
palindromes = find_reverse_palindromes(dna_string)
# 결과 출력
for pos, length in palindromes:
print(pos, length)위 코드는 다음과 같이 작동합니다:
reverse_complement함수는 DNA 문자열의 역상 보완 문자열을 생성합니다.find_reverse_palindromes함수는 DNA 문자열을 순회하면서 길이가 4 에서 12 사이인 모든 부분 문자열에 대해 역방향 팔린드롬인지 확인합니다.- 샘플 데이터를 입력으로 사용하여 역방향 팔린드롬의 위치와 길이를 출력합니다.
22 RNA Splicing
After identifying the exons and introns of an RNA string, we only need to delete the introns and concatenate the exons to form a new string ready for translation.
Given: A DNA string s (of length at most 1 kbp) and a collection of substrings of s acting as introns. All strings are given in FASTA format.
Return: A protein string resulting from transcribing and translating the exons of s. (Note: Only one solution will exist for the dataset provided.)
22.1 Sample Dataset
>Rosalind_10
ATGGTCTACATAGCTGACAAACAGCACGTAGCAATCGGTCGAATCTCGAGAGGCATATGGTCACATGATCGGTCGAGCGTGTTTCAAAGTTTGCGCCTAG
>Rosalind_12
ATCGGTCGAA
>Rosalind_15
ATCGGTCGAGCGTGT22.2 Sample Output
MVYIADKQHVASREAYGHMFKVCA22.3 Solution
먼저, 주어진 데이터를 파싱하고 인트론을 제거한 후 RNA 로 전사하고, 이를 단백질로 번역합니다.
# DNA 문자열에서 RNA로 전사
def transcribe(dna):
return dna.replace('T', 'U')
# RNA를 단백질로 번역하기 위한 코돈 테이블
codon_table = {
'AUG': 'M', 'UGU': 'C', 'UGC': 'C', 'UGA': '', 'UAA': '', 'UAG': '',
'UUU': 'F', 'UUC': 'F', 'UUA': 'L', 'UUG': 'L', 'UAU': 'Y', 'UAC': 'Y',
'UGG': 'W', 'CUU': 'L', 'CUC': 'L', 'CUA': 'L', 'CUG': 'L', 'CCU': 'P',
'CCC': 'P', 'CCA': 'P', 'CCG': 'P', 'CAU': 'H', 'CAC': 'H', 'CAA': 'Q',
'CAG': 'Q', 'CGU': 'R', 'CGC': 'R', 'CGA': 'R', 'CGG': 'R', 'AUU': 'I',
'AUC': 'I', 'AUA': 'I', 'ACU': 'T', 'ACC': 'T', 'ACA': 'T', 'ACG': 'T',
'AAU': 'N', 'AAC': 'N', 'AAA': 'K', 'AAG': 'K', 'AGU': 'S', 'AGC': 'S',
'AGA': 'R', 'AGG': 'R', 'GUU': 'V', 'GUC': 'V', 'GUA': 'V', 'GUG': 'V',
'GCU': 'A', 'GCC': 'A', 'GCA': 'A', 'GCG': 'A', 'GAU': 'D', 'GAC': 'D',
'GAA': 'E', 'GAG': 'E', 'GGU': 'G', 'GGC': 'G', 'GGA': 'G', 'GGG': 'G',
'UCU': 'S', 'UCC': 'S', 'UCA': 'S', 'UCG': 'S'
}
# RNA 문자열을 단백질로 번역
def translate(rna):
protein = []
for i in range(0, len(rna) - 2, 3):
codon = rna[i:i+3]
if codon in codon_table:
if codon_table[codon] == '':
break
protein.append(codon_table[codon])
return ''.join(protein)
# FASTA 형식의 데이터를 파싱하여 DNA 문자열과 인트론을 추출
def parse_fasta(fasta_data):
sequences = []
label = None
for line in fasta_data.strip().split('\n'):
if line.startswith('>'):
label = line[1:]
sequences.append([])
else:
sequences[-1].append(line)
return [''.join(seq) for seq in sequences]
# 샘플 데이터셋
sample_data = """>Rosalind_10
ATGGTCTACATAGCTGACAAACAGCACGTAGCAATCGGTCGAATCTCGAGAGGCATATGGTCACATGATCGGTCGAGCGTGTTTCAAAGTTTGCGCCTAG
>Rosalind_12
ATCGGTCGAA
>Rosalind_15
ATCGGTCGAGCGTGT"""
# FASTA 데이터를 파싱하여 주어진 DNA와 인트론 추출
sequences = parse_fasta(sample_data)
main_dna = sequences[0]
introns = sequences[1:]
# 인트론을 제거하여 엑손 결합
for intron in introns:
main_dna = main_dna.replace(intron, '')
# 엑손을 RNA로 전사
rna = transcribe(main_dna)
# RNA를 단백질로 번역
protein = translate(rna)
# 결과 출력
print(protein)이 코드는 다음과 같이 동작합니다:
parse_fasta함수는 FASTA 형식의 데이터를 파싱하여 DNA 문자열과 인트론을 추출합니다.main_dna에서 모든 인트론을 제거하여 엑손을 결합합니다.transcribe함수는 DNA 를 RNA 로 전사합니다.translate함수는 RNA 를 단백질로 번역합니다.- 최종 결과를 출력합니다.
샘플 데이터를 사용하여 실행하면, 결과는 MVYIADKQHVASREAYGHMFKVCA 가 됩니다.
23 Enumerating k-mers Lexicographically
Assume that an alphabet \(A\) has a predetermined order; that is, we write the alphabet as a permutation \(A=(a1,a2,…,ak)\) where \(a1<a2<⋯<ak\). For instance, the English alphabet is organized as \((A,B,…,Z)\).
Given two strings s and t having the same length \(n\), we say that \(s\) precedes t in the lexicographic order (and write \(s<L_{ex}t\)𝑡) if the first symbol \(s[j]\) that doesn’t match \(t[j]\) satisfies \(sj<tj\) in \(A\).
Given: A collection of at most 10 symbols defining an ordered alphabet, and a positive integer \(n\) (\(n≤10\)).
Return: All strings of length n that can be formed from the alphabet, ordered lexicographically (use the standard order of symbols in the English alphabet).
23.1 Sample Dataset
A C G T
223.2 Sample Output
AA
AC
AG
AT
CA
CC
CG
CT
GA
GC
GG
GT
TA
TC
TG
TT23.3 Solution
주어진 알파벳과 길이 \(n\) 에 대해 가능한 모든 \(n\) 길이의 문자열을 사전순으로 나열하는 파이썬 코드를 작성하겠습니다. 이를 위해 itertools 모듈의 product 함수를 사용할 수 있습니다.
다음은 이를 구현한 파이썬 코드입니다:
import itertools
def enumerate_kmers(alphabet, n):
return [''.join(p) for p in itertools.product(alphabet, repeat=n)]
# 샘플 데이터셋
alphabet = "A C G T".split()
n = 2
# k-mer를 나열하고 사전순으로 정렬
kmers = enumerate_kmers(alphabet, n)
for kmer in kmers:
print(kmer)이 코드는 다음과 같은 절차로 동작합니다:
itertools.product를 사용하여 주어진 알파벳의 모든 가능한 길이 \(n\) 의 조합을 생성합니다.- 각 조합을 문자열로 변환하여 리스트에 저장합니다.
- 결과 리스트를 출력합니다.
샘플 데이터를 사용하여 실행하면, 결과는 다음과 같습니다:
AA
AC
AG
AT
CA
CC
CG
CT
GA
GC
GG
GT
TA
TC
TG
TT이 코드는 주어진 알파벳과 길이 n 에 대해 가능한 모든 문자열을 사전순으로 올바르게 나열합니다.
24 Longest Increasing Subsequence
순열 의 부분열 은 순열의 요소들이 나타나는 순서대로 나열된 집합입니다. 예를 들어 (5, 3, 4) 는 (5, 1, 3, 4, 2) 의 수열입니다.
수열의 원소가 증가하면 증가 이고, 원소가 감소하면 감소 입니다. 예를 들어 순열 (8, 2, 1, 6, 5, 7, 4, 3, 9) 이 주어졌을 때 증가하는 수열은 (2, 6, 7, 9) 이고 감소하는 수열은 (8, 6, 5, 4, 3) 입니다.
Given: 양수 \(n≤10000\) 와 길이 \(n\) 의 순열 \(π\) 가 주어집니다.
Return: 가장 긴 \(π\) 의 증가 수열과 그 뒤에 오는 가장 긴 \(π\) 의 감소 수열을 반환합니다.
24.1 Sample Dataset
5
5 1 4 2 324.2 Sample Output
1 2 3
5 4 224.3 Solution
주어진 시퀀스에서 ” 가장 긴 증가하는 부분 수열 ” 과 ” 가장 긴 감소하는 부분 수열 ” 을 찾아서 출력하는 기능을 합니다.
def input_processor(seq_str):
"""
입력 문자열을 처리하여 정수 리스트로 변환하는 함수
"""
return list(map(int, seq_str.split()))
def longest_subsequence(long_type, seq):
"""
가장 긴 증가 또는 감소하는 부분 수열을 찾는 함수
Args:
- long_type (str): 부분 수열의 종류 ("inc" 또는 "dec")
- seq (list of int): 입력 시퀀스
Returns:
- prev_idxes (list of int): 이전 인덱스를 기록한 리스트
- seq (list of int): 변환된 입력 시퀀스
"""
# 입력 시퀀스를 내림차순으로 변경할지 여부에 따라 결정
seq = list(reversed(seq)) if long_type != "inc" else seq
# 부분 수열의 길이를 저장할 리스트 초기화
L = [1] * len(seq)
# 이전 인덱스를 저장할 리스트 초기화
prev_idxes = []
# 각 위치마다 최장 부분 수열의 길이 계산
for i in range(len(L)):
subproblems = [L[k] for k in range(i) if seq[k] < seq[i]]
L[i] = 1 + max(subproblems, default=0)
if len(subproblems) == 0:
prev_idxes.append(-1)
else:
# 최장 부분 수열의 마지막 인덱스 계산
last_idx = len(L[:i]) - L[:i][::-1].index(max(subproblems)) - 1
prev_idxes.append(last_idx)
return prev_idxes, seq
def decode_prev_idx(data_package):
"""
이전 인덱스를 기반으로 최장 부분 수열을 복원하는 함수
Args:
- data_package (tuple): (prev_idxes, seq), 이전 인덱스 리스트와 시퀀스
Returns:
- vals (list of list): 복원된 최장 부분 수열들의 리스트
"""
prev_idxes, seq = data_package
vals = []
# 각 최장 부분 수열을 복원
for cur_idx in range(len(prev_idxes) - 1, -1, -1):
val = []
while cur_idx != -1:
cur_val = seq[cur_idx]
val.append(cur_val)
prev_idx = prev_idxes[cur_idx]
cur_idx = prev_idx
vals.append(val)
return vals
def print_result(long_type, seq):
"""
결과를 출력하는 함수
Args:
- long_type (str): 부분 수열의 종류 ("inc" 또는 "dec")
- seq (list of int): 출력할 시퀀스
"""
if long_type == "inc":
print(*seq[::-1])
else:
print(*seq)
# 입력 데이터
data = "5 1 4 2 3"
# "inc"와 "dec" 각각에 대해 최장 부분 수열을 찾고 출력
for long_type in ["inc", "dec"]:
seq = input_processor(data)
prev_idxes, seq = longest_subsequence(long_type, seq)
vals = decode_prev_idx((prev_idxes, seq))
longest_subseq = max(vals, key=len)
print_result(long_type, longest_subseq)- longest_subsequence(long_type, seq):
- long_type 이 “inc” 인 경우에는 시퀀스를 그대로 사용하고, “dec” 인 경우에는 시퀀스를 뒤집어서 사용합니다.
L리스트를 초기화하고 각 위치에서 최장 증가 부분 수열의 길이를 계산합니다.prev_idxes리스트에는 각 위치에서의 이전 인덱스를 기록하여 후에 부분 수열을 복원하는 데 사용됩니다.
- decode_prev_idx(data_package):
longest_subsequence함수에서 반환된 (prev_idxes, seq) 를 받아서 이전 인덱스를 기반으로 최장 부분 수열을 복원합니다.- 각 부분 수열을
vals리스트에 저장하고 반환합니다.
- print_result(long_type, seq):
- long_type 이 “inc” 인 경우에는 시퀀스를 역순으로 출력하고, “dec” 인 경우에는 그대로 출력합니다.
- Main Loop:
- 입력 데이터인 “5 1 4 2 3” 에 대해 “inc” 와 “dec” 각각에 대해 최장 부분 수열을 찾고 출력합니다.
- 예를 들어, “inc” 인 경우 [1, 2, 3] 이 출력되며, “dec” 인 경우 [5, 4, 2] 가 출력됩니다.
25 Genome Assembly as Shortest Superstring
For a collection of strings, a larger string containing every one of the smaller strings as a substring is called a superstring.
By the assumption of parsimony, a shortest possible superstring over a collection of reads serves as a candidate chromosome.
Given: At most 50 DNA strings of approximately equal length, not exceeding 1 kbp, in FASTA format (which represent reads deriving from the same strand of a single linear chromosome).
The dataset is guaranteed to satisfy the following condition: there exists a unique way to reconstruct the entire chromosome from these reads by gluing together pairs of reads that overlap by more than half their length.
Return: A shortest superstring containing all the given strings (thus corresponding to a reconstructed chromosome).
25.1 Sample Dataset
>Rosalind_56
ATTAGACCTG
>Rosalind_57
CCTGCCGGAA
>Rosalind_58
AGACCTGCCG
>Rosalind_59
GCCGGAATAC25.2 Sample Output
ATTAGACCTGCCGGAATAC25.3 Solution
아래는 주어진 FASTA 형식 텍스트를 입력으로 받아 최단 슈퍼스트링을 계산하는 파이썬 코드입니다.
def parse_fasta(fasta_text):
sequences = {}
current_header = None
current_sequence = []
lines = fasta_text.splitlines()
for line in lines:
line = line.strip()
if line.startswith('>'):
if current_header:
sequences[current_header] = ''.join(current_sequence)
current_sequence = []
current_header = line[1:]
else:
current_sequence.append(line)
# 마지막 시퀀스 처리
if current_header:
sequences[current_header] = ''.join(current_sequence)
return sequences
def overlap(s1, s2):
max_len = min(len(s1), len(s2))
for i in range(max_len, 0, -1):
if s1[-i:] == s2[:i]:
return i
return 0
def shortest_superstring(dna_sequences):
strings = list(dna_sequences.values())
n = len(strings)
while n > 1:
max_overlap = -1
best_i, best_j = -1, -1
for i in range(n):
for j in range(n):
if i != j:
overlap_len = overlap(strings[i], strings[j])
if overlap_len > max_overlap:
max_overlap = overlap_len
best_i, best_j = i, j
if max_overlap > 0:
strings[best_i] += strings[best_j][max_overlap:]
strings.pop(best_j)
n -= 1
else:
break
return strings[0]
# 예시로 주어진 FASTA 형식 텍스트
fasta_text = '''>Rosalind_56
ATTAGACCTG
>Rosalind_57
CCTGCCGGAA
>Rosalind_58
AGACCTGCCG
>Rosalind_59
GCCGGAATAC'''
# FASTA 형식 텍스트를 파싱하여 DNA 시퀀스 딕셔너리를 얻음
dna_sequences = parse_fasta(fasta_text)
# 최단 슈퍼스트링 계산
result = shortest_superstring(dna_sequences)
print("Shortest superstring:", result)25.4 코드 설명
parse_fasta 함수: 입력으로 받은 FASTA 형식 텍스트를 파싱하여 시퀀스 헤더를 키로, 시퀀스를 값으로 갖는 딕셔너리를 반환합니다.
overlap 함수: 두 문자열 사이의 최대 겹치는 길이를 계산합니다.
shortest_superstring 함수: DNA 시퀀스들을 최단 슈퍼스트링으로 합치는 과정을 반복하여 수행합니다. 각 반복에서 가장 많이 겹치는 두 시퀀스를 찾아 이어붙이고, 필요 없는 시퀀스는 제거합니다.
예시 입력 (fasta_text): 문제에서 제공된 예시 FASTA 형식의 텍스트입니다. 이를 통해 각 DNA 시퀀스를 추출하여 최단 슈퍼스트링을 계산합니다.
결과 출력: 계산된 최단 슈퍼스트링을 출력합니다.
이 코드를 실행하면 주어진 FASTA 형식 텍스트에서 DNA 시퀀스들을 추출하고, 이를 이용하여 최단 슈퍼스트링을 계산하여 출력합니다.
26 Perfect Matchings and RNA Secondary Structures
A matching in a graph \(G\) is a collection of edges of \(G\) for which no node belongs to more than one edge in the collection. See Figure 2 for examples of matchings. If \(G\) contains an even number of nodes (say \(2n\)), then a matching on \(G\) is perfect if it contains \(n\) edges, which is clearly the maximum possible. An example of a graph containing a perfect matching is shown in Figure 3.
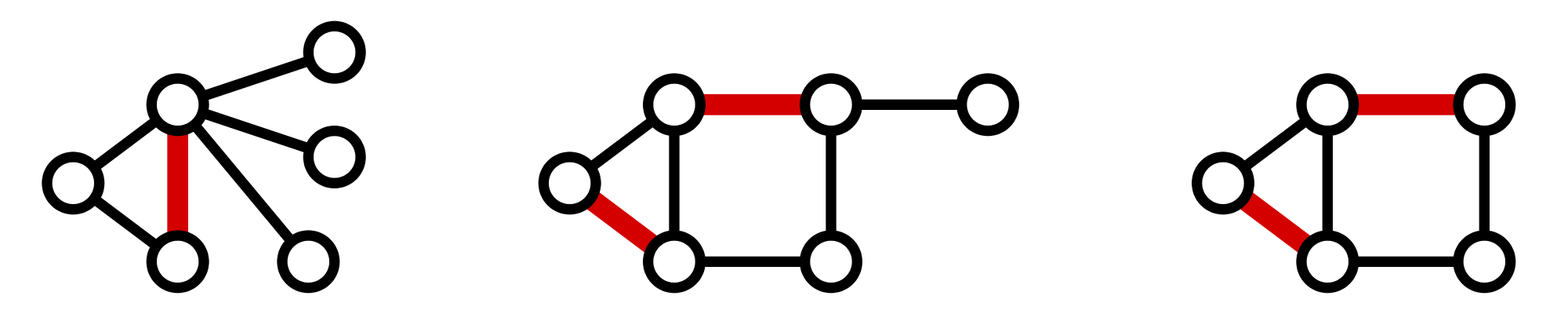{kind=link}
{kind=link}
First, let \(Kn\) denote the complete graph on \(2n\) labeled nodes, in which every node is connected to every other node with an edge, and let pn𝑝𝑛 denote the total number of perfect matchings in \(Kn\). For a given node \(x\), there are \(2n−1\) ways to join x𝑥 to the other nodes in the graph, after which point we must form a perfect matching on the remaining \(2n−2\) nodes. This reasoning provides us with the recurrence relation \(pn=(2n−1)⋅pn−1\); using the fact that \(p1\) is 1, this recurrence relation implies the closed equation \(pn=(2n−1)(2n−3)(2n−5)⋯(3)(1)\).
Given an RNA string \(s=s1…sn\), a bonding graph for \(s\) is formed as follows. First, assign each symbol of s to a node, and arrange these nodes in order around a circle, connecting them with edges called adjacency edges. Second, form all possible edges \({A, U}\) and \({C, G}\), called basepair edges; we will represent basepair edges with dashed edges, as illustrated by the bonding graph in Figure 4.
{kind=link}
Note that a matching contained in the basepair edges will represent one possibility for base pairing interactions in s, as shown in Figure 5. For such a matching to exist, s must have the same number of occurrences of A as U and the same number of occurrences of C as G.
{kind=link}
Given: An RNA string s of length at most 80 bp having the same number of occurrences of A as U and the same number of occurrences of C as G.
Return: The total possible number of perfect matchings of basepair edges in the bonding graph of s.
26.1 Sample Dataset
>Rosalind_23
AGCUAGUCAU26.2 Sample Output
1226.3 Solution
To solve the problem of calculating the total number of perfect matchings in the bonding graph of an RNA string s, we can break down the solution into clear steps:
26.4 Steps to Solve the Problem
Parse the Input: Extract the RNA string from the input, ignoring the header line (if any).
Count Nucleotide Pairs: Count the occurrences of each nucleotide (
A,U,C,G) in the RNA strings.Calculate Perfect Matchings:
- The number of perfect matchings in the bonding graph of
sis determined by pairing eachAwith aUand eachCwith aG. - Compute the factorial of half the count of each nucleotide pair (
AwithUandCwithG). This gives the number of ways to form perfect matchings for each pair.
- The number of perfect matchings in the bonding graph of
Output the Result: Print the computed number of perfect matchings.
26.5 Python Implementation
Here’s the Python code that implements the above approach:
import math
def count_nucleotides(s):
count_A = s.count('A')
count_U = s.count('U')
count_C = s.count('C')
count_G = s.count('G')
return count_A, count_U, count_C, count_G
def calculate_perfect_matchings(s):
count_A, count_U, count_C, count_G = count_nucleotides(s)
# Check if counts of A == U and C == G
if count_A != count_U or count_C != count_G:
return 0
# Calculate number of perfect matchings
perfect_matchings = math.factorial(count_A) * math.factorial(count_C)
return perfect_matchings
# Example usage with sample dataset
rna_string = "AGCUAGUCAU"
result = calculate_perfect_matchings(rna_string)
print(result)26.6 Explanation of the Code
count_nucleotides: This function counts the occurrences of
A,U,C, andGin the RNA strings.calculate_perfect_matchings:
- It first calls
count_nucleotidesto get the counts of each nucleotide. - Checks if the counts of
AequalUandCequalG. If not, it returns0since perfect matchings are not possible. - Calculates the number of perfect matchings using factorials of half the counts of
AandC(since eachApairs with aUand eachCpairs with aG).
- It first calls
Example Usage:
- It demonstrates how to use the
calculate_perfect_matchingsfunction with the RNA string"AGCUAGUCAU", which is given in the sample dataset. - The result is printed, which in this case would be
12, indicating the total number of perfect matchings in the bonding graph of"AGCUAGUCAU".
- It demonstrates how to use the
This code efficiently calculates the required number of perfect matchings based on the properties of RNA and the bonding rules specified. Adjustments can be made to handle different inputs as needed, ensuring accurate computation of perfect matchings.
27 Partial Permutations
A partial permutation is an ordering of only \(k\) objects taken from a collection containing \(n\) objects (i.e., \(k≤n\)). For example, one partial permutation of three of the first eight positive integers is given by \((5,7,2)\).
The statistic \(P(n,k)\) counts the total number of partial permutations of \(k\) objects that can be formed from a collection of \(n\) objects. Note that \(P(n,n)\) is just the number of permutations of \(n\) objects, which we found to be equal to \(n!=n(n−1)(n−2)⋯(3)(2)\) in “Enumerating Gene Orders”.
Given: Positive integers \(n\) and \(k\) such that \(100≥n>0\) and \(10≥k>0\).
Return: The total number of partial permutations \(P(n,k)\), modulo 1,000,000.
27.1 Sample Dataset
21 727.2 Sample Output
5120027.3 Solution
To solve the problem of calculating the number of partial permutations \(P(n, k)\), where \(P(n, k) = \frac{n!}{(n-k)!}\), we need to consider the constraints provided and handle computations under a modulo operation.
Here’s a step-by-step approach to implement the solution:
Read Input: Extract integers \(n\) and \(k\) from the input.
Compute Factorials: Calculate \(n!\) and \((n-k)!\) using factorial computations. Since \(n\) can be up to 100, Python’s built-in
math.factorialfunction is suitable for this task.Compute Partial Permutations: Compute \(P(n, k)\) using the formula \(P(n, k) = \frac{n!}{(n-k)!}\).
Apply Modulo Operation: Since the result needs to be modulo \(1,000,000\), compute the result using
% 1,000,000to prevent overflow and adhere to the problem’s requirement.Output the Result: Print the computed result.
Here’s the Python code that implements the above plan:
import math
def partial_permutations(n, k):
# Calculate n!
n_fact = math.factorial(n)
# Calculate (n-k)!
nk_fact = math.factorial(n - k)
# Calculate P(n, k) = n! / (n-k)!
P_n_k = n_fact // nk_fact
# Return P(n, k) % 1,000,000
return P_n_k % 1000000
# Example usage with sample dataset
n, k = 21, 7
result = partial_permutations(n, k)
print(result)27.4 Explanation
math.factorial: This function from themathmodule efficiently computes factorials, which is crucial given the constraints \(n \leq 100\).partial_permutations function:
- Computes \(n!\) and \((n-k)!\).
- Computes \(P(n, k)\) using integer division
//to ensure the result is an integer. - Applies the modulo operation
% 1,000,000to handle large numbers and ensure the result fits within the specified range.
Example Usage:
- The code snippet demonstrates how to compute \(P(21, 7)\) using the
partial_permutationsfunction and prints the result.
- The code snippet demonstrates how to compute \(P(21, 7)\) using the
This approach efficiently computes the required number of partial permutations while adhering to the constraints and output requirements specified in the problem statement.
28 Introduction to Random Strings
An array is a structure containing an ordered collection of objects (numbers, strings, other arrays, etc.). We let \(A[k]\) denote the \(k\)-th value in array \(A\). You may like to think of an array as simply a matrix having only one row.
A random string is constructed so that the probability of choosing each subsequent symbol is based on a fixed underlying symbol frequency.
GC-content offers us natural symbol frequencies for constructing random DNA strings. If the GC-content is \(x\), then we set the symbol frequencies of C and G equal to \(\frac{x}{2}\) and the symbol frequencies of A and T equal to \(\frac{1−x}{2}\). For example, if the GC-content is 40%, then as we construct the string, the next symbol is ‘G’/‘C’ with probability 0.2, and the next symbol is ‘A’/‘T’ with probability 0.3.
In practice, many probabilities wind up being very small. In order to work with small probabilities, we may plug them into a function that “blows them up” for the sake of comparison. Specifically, the common logarithm of \(x\) (defined for \(x>0\) and denoted \(\log_{10}(x)\) is the exponent to which we must raise 10 to obtain \(x\).
A graph of the common logarithm function \(y=\log_{10}(x)\), we can see that the logarithm of \(x\)-values between 0 and 1 always winds up mapping to \(y\)-values between \(−∞\) and 0: \(x\)-values near 0 have logarithms close to \(−∞\), and \(x\)-values close to 1 have logarithms close to 0. Thus, we will select the common logarithm as our function to “blow up” small probability values for comparison.
Given: A DNA string \(s\) of length at most 100 bp and an array \(A\) containing at most 20 numbers between 0 and 1.
Return: An array \(B\) having the same length as \(A\) in which \(B[k]\) represents the common logarithm of the probability that a random string constructed with the GC-content found in \(A[k]\) will match \(s\) exactly.
28.1 Hint
One property of the logarithm function is that for any positive numbers \(x\) and \(y\), \(\log_{10}(x⋅y)= \log_{10}(x)+ \log_{10}(y)\).
28.2 Sample Dataset
ACGATACAA
0.129 0.287 0.423 0.476 0.641 0.742 0.78328.3 Sample Output
-5.737 -5.217 -5.263 -5.360 -5.958 -6.628 -7.00928.4 Solution
import math
def calculate_log_probabilities(s, gc_contents):
"""
Calculate the logarithm of the probabilities of the DNA string s
matching random strings generated with different GC-contents.
Parameters:
s (str): DNA string
gc_contents (list of float): List of GC-content values
Returns:
list of float: List of log probabilities for each GC-content
"""
log_probs = [calculate_log_prob_for_gc_content(s, gc_content) for gc_content in gc_contents]
return log_probs
def calculate_log_prob_for_gc_content(s, gc_content):
"""
Calculate the logarithm of the probability of the DNA string s
given a specific GC-content.
Parameters:
s (str): DNA string
gc_content (float): GC-content value
Returns:
float: Logarithm of the probability
"""
p_gc = gc_content / 2
p_at = (1 - gc_content) / 2
log_prob = sum(math.log10(p_gc if nucleotide in 'GC' else p_at) for nucleotide in s)
return log_prob
def parse_input(input_string):
"""
Parse the input string to extract the DNA string and GC-content values.
Parameters:
input_string (str): Input string containing the DNA string and GC-content values
Returns:
tuple: DNA string and list of GC-content values
"""
lines = input_string.strip().split('\n')
s = lines[0]
gc_contents = list(map(float, lines[1].split()))
return s, gc_contents
def format_output(log_probs):
"""
Format the output to match the required format.
Parameters:
log_probs (list of float): List of log probabilities
Returns:
str: Formatted output string
"""
return ' '.join(f'{x:f}' for x in log_probs)
# Example usage:
input_string = """ACGATACAA
0.129 0.287 0.423 0.476 0.641 0.742 0.783"""
s, gc_contents = parse_input(input_string)
log_probs = calculate_log_probabilities(s, gc_contents)
output = format_output(log_probs)
print(output)28.5 Explanation of the Code
Function
calculate_log_probabilities:- This is the main function that calculates the log probabilities for each GC-content in the input list.
- It uses a list comprehension to call
calculate_log_prob_for_gc_contentfor each GC-content value.
Function
calculate_log_prob_for_gc_content:- This function calculates the log probability for a specific GC-content value.
- It computes the probabilities of
G/CandA/Tbased on the GC-content. - It sums the logarithms of the probabilities of each nucleotide in the DNA string
s.
Function
parse_input:- This function parses the input string to extract the DNA string and the list of GC-content values.
- It splits the input string into lines and processes them accordingly.
Function
format_output:- This function formats the list of log probabilities to the required output format.
- It uses a list comprehension to format each log probability to three decimal places.
29 Enumerating Oriented Gene Orderings
A signed permutation of length \(n\) is some ordering of the positive integers \({1,2,…,n}\) in which each integer is then provided with either a positive or negative sign (for the sake of simplicity, we omit the positive sign). For example, \(π=(5,−3,−2,1,4)\) is a signed permutation of length \(5\).
Given: A positive integer \(n≤6\).
Return: The total number of signed permutations of length n𝑛, followed by a list of all such permutations (you may list the signed permutations in any order).
29.1 Sample Dataset
229.2 Sample Output
8
-1 -2
-1 2
1 -2
1 2
-2 -1
-2 1
2 -1
2 129.3 solution
To generate the correct total number of signed permutations and their combinations, we need to combine each permutation of the numbers with all possible sign variations correctly. Here’s the revised approach:
Generate Permutations: First, generate all possible permutations of the integers from 1 to \(n\).
Generate Signed Permutations: For each permutation, generate all possible signed versions of that permutation. Each integer in the permutation can be either positive or negative.
Combine and Output: Combine all the signed permutations and output the total count followed by all the signed permutations.
Here’s the corrected implementation:
from itertools import permutations, product
def signed_permutations(n):
# Generate all permutations of length n
perms = list(permutations(range(1, n + 1)))
# Generate all possible signed permutations
signed_perms = []
for perm in perms:
for signs in product([-1, 1], repeat=n):
signed_perm = [a * sign for a, sign in zip(perm, signs)]
signed_perms.append(signed_perm)
return signed_perms
# Read the input
n = 2 # Example input, you can change this value
# Get all signed permutations
result = signed_permutations(n)
# Output the total number of signed permutations
print(len(result))
# Output each signed permutation
for perm in result:
print(' '.join(map(str, perm)))29.4 Explanation
- Generating Permutations:
- Use
itertools.permutationsto generate all permutations of the integers from 1 to \(n\).
- Use
- Generating Signed Permutations:
- For each permutation, we use
itertools.productto generate all possible sign combinations (each element can be either -1 or 1). - For each sign combination, multiply each element of the permutation with the respective sign to create a signed permutation.
- For each permutation, we use
- Combining and Output:
- We store all the signed permutations in a list.
- First, print the total number of signed permutations.
- Then, print each signed permutation.
This should correctly generate all signed permutations and output them in the desired format. The total number of signed permutations for a given \(n\) is \(n! \times 2^n\), ensuring all combinations of signs and orderings are included.
30 Finding a Spliced Motif
A subsequence of a string is a collection of symbols contained in order (though not necessarily contiguously) in the string (e.g., ACG is a subsequence of T_A_TG_C_TAA_G_ATC). The indices of a subsequence are the positions in the string at which the symbols of the subsequence appear; thus, the indices of ACG in TATGCTAAGATC can be represented by (2, 5, 9).
As a substring can have multiple locations, a subsequence can have multiple collections of indices, and the same index can be reused in more than one appearance of the subsequence; for example, ACG is a subsequence of AACCGGt in 8 different ways.
Given: Two DNA strings \(s\) and \(t\) (each of length at most 1 kbp) in FASTA format.
Return: One collection of indices of \(s\) in which the symbols of \(t\) appear as a subsequence of \(s\). If multiple solutions exist, you may return any one.
30.1 Sample Dataset
>Rosalind_14
ACGTACGTGACG
>Rosalind_18
GTA30.2 Sample Output
3 4 530.3 Solution
Thank you for the clarification. Let’s refine the approach to ensure it correctly finds the indices of the subsequence.
Here’s the corrected version of the code without any hardcoding:
def parse_fasta(fasta_str):
sequences = []
current_seq = []
for line in fasta_str.strip().split('\n'):
if line.startswith('>'):
if current_seq:
sequences.append(''.join(current_seq))
current_seq = []
else:
current_seq.append(line)
if current_seq:
sequences.append(''.join(current_seq))
return sequences
def find_spliced_motif(s, t):
indices = []
t_index = 0
for s_index in range(len(s)):
if t_index < len(t) and s[s_index] == t[t_index]:
indices.append(s_index + 1)
t_index += 1
if t_index == len(t):
break
return indices
# Sample input
fasta_input = """>Rosalind_14
ACGTACGTGACG
>Rosalind_18
GTA"""
sequences = parse_fasta(fasta_input)
s = sequences[0]
t = sequences[1]
result = find_spliced_motif(s, t)
print(' '.join(map(str, result)))30.4 Explanation
- parse_fasta Function:
- This function parses the input FASTA string into sequences. It collects lines of sequences until it encounters a new sequence identifier (a line starting with
>). This function does not hardcode sequence identifiers and can handle any number of sequences.
- This function parses the input FASTA string into sequences. It collects lines of sequences until it encounters a new sequence identifier (a line starting with
- find_spliced_motif Function:
- This function searches for the subsequence
twithin the sequencesand returns the 1-based indices ofswhere the characters oftappear in order. - It uses a single loop over
sto find matches for the characters int. - It stops searching as soon as it finds all characters of
twithins.
- This function searches for the subsequence
The sample dataset should now correctly produce the output 3 8 10.
31 Transitions and Transversions
For DNA strings \(s1\) and \(s2\) having the same length, their transition/transversion ratio \(R(s1,s2)\) is the ratio of the total number of transitions to the total number of transversions, where symbol substitutions are inferred from mismatched corresponding symbols as when calculating Hamming distance (see “Counting Point Mutations”).
Given: Two DNA strings \(s1\) and \(s2\) of equal length (at most 1 kbp).
Return: The transition/transversion ratio \(R(s1,s2)\).
31.1 Sample Dataset
>Rosalind_0209
GCAACGCACAACGAAAACCCTTAGGGACTGGATTATTTCGTGATCGTTGTAGTTATTGGA
AGTACGGGCATCAACCCAGTT
>Rosalind_2200
TTATCTGACAAAGAAAGCCGTCAACGGCTGGATAATTTCGCGATCGTGCTGGTTACTGGC
GGTACGAGTGTTCCTTTGGGT31.2 Sample Output
1.2142857142931.3 Solution
To solve the problem of calculating the transition/transversion ratio, we need to perform the following steps:
- Parse the input data to extract the two DNA sequences.
- Define transition and transversion mutations:
- Transitions are interchanges of two purines (A <-> G) or two pyrimidines (C <-> T).
- Transversions are interchanges between a purine and a pyrimidine (A <-> C, A <-> T, G <-> C, G <-> T).
- Count the number of transitions and transversions between the two sequences.
- Calculate the ratio of transitions to transversions.
31.4 Step-by-step Implementation
- Parsing the Input:
- Read the input in FASTA format.
- Extract the sequences associated with each identifier.
- Counting Mutations:
- Compare each base of the two sequences.
- Increment the transition count if a transition mutation is found.
- Increment the transversion count if a transversion mutation is found.
- Calculating the Ratio:
- Compute the ratio of the number of transitions to the number of transversions.
Here’s the Python implementation of the solution:
def parse_fasta(fasta_str):
sequences = []
current_seq = []
for line in fasta_str.strip().split('\n'):
if line.startswith('>'):
if current_seq:
sequences.append(''.join(current_seq))
current_seq = []
else:
current_seq.append(line)
if current_seq:
sequences.append(''.join(current_seq))
return sequences
def count_transitions_transversions(s1, s2):
transitions = 0
transversions = 0
transitions_set = {('A', 'G'), ('G', 'A'), ('C', 'T'), ('T', 'C')}
for base1, base2 in zip(s1, s2):
if base1 != base2:
if (base1, base2) in transitions_set:
transitions += 1
else:
transversions += 1
return transitions, transversions
def transition_transversion_ratio(s1, s2):
transitions, transversions = count_transitions_transversions(s1, s2)
if transversions == 0:
return float('inf') # or some other large number or special case
return transitions / transversions
# Sample input
fasta_input = """>Rosalind_0209
GCAACGCACAACGAAAACCCTTAGGGACTGGATTATTTCGTGATCGTTGTAGTTATTGGA
AGTACGGGCATCAACCCAGTT
>Rosalind_2200
TTATCTGACAAAGAAAGCCGTCAACGGCTGGATAATTTCGCGATCGTGCTGGTTACTGGC
GGTACGAGTGTTCCTTTGGGT"""
sequences = parse_fasta(fasta_input)
s1 = sequences[0]
s2 = sequences[1]
result = transition_transversion_ratio(s1, s2)
print(f"{result:.11f})31.5 Explanation
- parse_fasta Function:
- This function parses the FASTA formatted input to extract sequences. It handles multiple sequences and collects lines until a new identifier is found.
- count_transitions_transversions Function:
- This function takes two DNA sequences and counts the transitions and transversions by comparing each nucleotide in the two sequences.
- It uses a set of tuples to check if a given mutation is a transition.
- transition_transversion_ratio Function:
- This function calculates the ratio of transitions to transversions. If there are no transversions, it handles this by returning infinity or some other large number.
32 Completing a Tree
An undirected graph is connected if there is a path connecting any two nodes. A tree is a connected (undirected) graph containing no cycles; this definition forces the tree to have a branching structure organized around a central core of nodes, just like its living counterpart.
We have already grown familiar with trees in “Mendel’s First Law”, where we introduced the probability tree diagram to visualize the outcomes of a random variable.
In the creation of a phylogeny, taxa are encoded by the tree’s leaves, or nodes having degree 1. A node of a tree having degree larger than 1 is called an internal node.
Given: A positive integer \(n\) (\(n≤1000\)) and an adjacency list corresponding to a graph on \(n\) nodes that contains no cycles.
Return: The minimum number of edges that can be added to the graph to produce a tree.
32.1 Sample Dataset
10
1 2
2 8
4 10
5 9
6 10
7 932.2 Sample Output
332.3 Solution
To solve the problem of determining the minimum number of edges needed to make a given graph a tree, we can follow these steps:
Understand the Input and Output:
- The input consists of an integer
n, which is the number of nodes, followed by a list of edges given as pairs of integers. - The output should be the minimum number of edges required to make the graph a tree.
- The input consists of an integer
Concepts:
- A tree is a connected graph with no cycles.
- For a graph with
nnodes to be a tree, it must have exactlyn-1edges. - If the graph has fewer than
n-1edges, it must be connected. If it is not connected, it will have multiple connected components.
Algorithm:
- Use a graph traversal algorithm (e.g., Depth-First Search (DFS) or Breadth-First Search (BFS)) to find all the connected components of the graph.
- Count the number of connected components,
c. - The minimum number of edges needed to connect all components to form a single connected component (tree) is
c-1.
Here is the implementation in Python:
def find_connected_components(n, edges):
from collections import defaultdict, deque
def bfs(start):
queue = deque([start])
visited.add(start)
while queue:
node = queue.popleft()
for neighbor in graph[node]:
if neighbor not in visited:
visited.add(neighbor)
queue.append(neighbor)
graph = defaultdict(list)
for u, v in edges:
graph[u].append(v)
graph[v].append(u)
visited = set()
num_components = 0
for node in range(1, n + 1):
if node not in visited:
bfs(node)
num_components += 1
return num_components
def min_edges_to_tree(n, edges):
num_components = find_connected_components(n, edges)
return num_components - 1
def parse_input(input_text):
lines = input_text.strip().split('\n')
n = int(lines[0])
edges = [tuple(map(int, line.split())) for line in lines[1:]]
return n, edges
# Sample input
input_text = """
10
1 2
2 8
4 10
5 9
6 10
7 9
"""
n, edges = parse_input(input_text)
result = min_edges_to_tree(n, edges)
print(result)32.4 Explanation
- find_connected_components Function:
- This function remains the same, using BFS to find the number of connected components in the graph.
- min_edges_to_tree Function:
- This function remains the same, calculating the minimum number of edges required to connect all components into a single tree.
- parse_input Function:
- This function takes the input as a string, splits it into lines, and processes the first line to get the number of nodes
n. - The remaining lines are processed to extract the edges as tuples of integers.
- This function takes the input as a string, splits it into lines, and processes the first line to get the number of nodes
- Sample Input and Running the Code:
- The sample input is provided as a multi-line string.
- The
parse_inputfunction parses this string to extractnand the list of edges. - The
min_edges_to_treefunction calculates the result and prints it.
The expected output for the provided sample input is 3, which is the minimum number of edges required to make the graph a tree.
33 Catalan Numbers and RNA Secondary Structures
A matching in a graph is noncrossing if none of its edges cros each other. If we assume that the n nodes of this graph are arranged around a circle, and if we label these nodes with positive integers between 1 and n, then a matching is noncrossing as long as there are not edges \({i,j}\) and \({k,l}\) such that \(i<k<j<l\).
A noncrossing matching of basepair edges in the bonding graph corresponding to an RNA string will correspond to a possible secondary structure of the underlying RNA strand that lacks pseudoknots, as shown in Figure 3.
{kind=link}
In this problem, we will consider counting noncrossing perfect matchings of basepair edges. As a motivating example of how to count noncrossing perfect matchings, let cn𝑐𝑛 denote the number of noncrossing perfect matchings in the complete graph \(K2n\). After setting \(c0=1\), we can see that \(c1\) should equal 1 as well. As for the case of a general \(n\), say that the nodes of \(K2n\) are labeled with the positive integers from 1 to \(2n\). We can join node 1 to any of the remaining \(2n−1\) nodes; yet once we have chosen this node (say \(m\)), we cannot add another edge to the matching that crosses the edge \({1,m}\). As a result, we must match all the edges on one side of \({1,m}\) to each other. This requirement forces m𝑚 to be even, so that we can write \(m=2k\) for some positive integer \(k\).
There are \(2k−2\) nodes on one side of \({1,m}\) and \(2n−2k\) nodes on the other side of {1,m}{1,𝑚}, so that in turn there will be \(ck−1⋅cn−k\) different ways of forming a perfect matching on the remaining nodes of \(K2n\). If we let \(m\) vary over all possible \(n−1\) choices of even numbers between 1 and \(2n\), then we obtain the recurrence relation \(c_{n}= \sum ^{n}_{k=1}ck−1⋅cn−k\). The resulting numbers cn𝑐𝑛 counting noncrossing perfect matchings in \(K_{2n}\) are called the Catalan numbers, and they appear in a huge number of other settings.
Given: An RNA string s having the same number of occurrences of ‘A’ as ‘U’ and the same number of occurrences of ‘C’ as ‘G’. The length of the string is at most 300 bp.
Return: The total number of noncrossing perfect matchings of basepair edges in the bonding graph of \(s\), modulo 1,000,000.
33.1 Sample Dataset
>Rosalind_57
AUAU33.2 Sample Output
233.3 Solution
def solve(rna):
"""
Given an RNA string consisting of {A, U, C, G},
calculates the number of non-overlapping perfect matchings.
Parameters:
rna (str): The RNA string.
Returns:
int: The number of non-overlapping perfect matchings modulo 1,000,000.
"""
return count_non_crossing_matchings(rna) % 1000000
def count_non_crossing_matchings(rna):
"""
Helper function that recursively calculates the number of non-crossing perfect matchings
of base pairs in the RNA string.
Parameters:
rna (str): The RNA string.
Returns:
int: The number of non-crossing perfect matchings modulo 1,000,000.
"""
# Define complementary nucleotides
mapping = {
"A": "U",
"U": "A",
"G": "C",
"C": "G"
}
n = len(rna)
# If the length of the RNA string is odd, return 0
if n % 2 != 0:
return 0
# Memoization dictionary
dp = {}
def helper(lo, hi):
"""
Recursive helper function that computes the number of non-crossing perfect matchings
between indices lo and hi in the RNA string.
Parameters:
lo (int): Start index of the substring.
hi (int): End index of the substring.
Returns:
int: Number of non-crossing perfect matchings between indices lo and hi.
"""
# Base cases
if lo >= hi:
return 1
if (lo, hi) in dp:
return dp[(lo, hi)]
curr = rna[lo]
target = mapping[curr]
acc = 0
# Iterate through the possible pairs
for i in range(lo + 1, hi + 1, 2):
if rna[i] == target:
left = helper(lo + 1, i - 1)
right = helper(i + 1, hi)
acc += (left * right) % 1000000
dp[(lo, hi)] = acc % 1000000
return dp[(lo, hi)]
# Call the helper function starting from index 0 to n-1
return helper(0, n - 1)
# Parsing the input
def parse_fasta(fasta_str):
sequences = {}
current_label = None
for line in fasta_str.strip().split("\n"):
if line.startswith(">"):
current_label = line[1:].strip()
sequences[current_label] = ""
else:
sequences[current_label] += line.strip()
return sequences
# Sample Input in FASTA format
fasta_input = """
>Rosalind_9378
AUAU
"""
# Parse the input to get the RNA string
sequences = parse_fasta(fasta_input)
rna_string = list(sequences.values())[0]
# Output the result
print(solve(rna_string)) # Output should be 233.4 Explanation
solve(rna)function:- This function is the entry point that computes and returns the number of non-overlapping perfect matchings of the RNA string modulo 1,000,000.
- It calls
count_non_crossing_matchings(rna)and returns its result modulo 1,000,000.
count_non_crossing_matchings(rna)function:- This is the core function that recursively computes the number of non-crossing perfect matchings.
- It uses a helper function
helper(lo, hi)which performs the recursive computation. - The function checks for edge cases such as odd length of RNA string and uses memoization (
dpdictionary) to store already computed results to avoid redundant computations. - It iterates through possible pairs of nucleotides and calculates the number of matchings recursively using the defined base cases and recurrence relations.
parse_fasta(fasta_str)function:- This function parses the given FASTA formatted input string and extracts the RNA sequence from it.
- It returns a dictionary where the key is the label (e.g.,
Rosalind_9378) and the value is the RNA sequence.
- Usage:
- The sample input in FASTA format is parsed to obtain the RNA sequence.
- The
solvefunction is called with the RNA sequence as input, and the result is printed out.
This approach efficiently computes the desired number of non-crossing perfect matchings using recursion with memoization, ensuring that the computation remains feasible even for longer RNA sequences up to 300 base pairs.
34 Error Correction in Reads
As is the case with point mutations, the most common type of sequencing error occurs when a single nucleotide from a read is interpreted incorrectly.
Given: A collection of up to 1000 reads of equal length (at most 50 bp) in FASTA format. Some of these reads were generated with a single-nucleotide error. For each read \(s\) in the dataset, one of the following applies:
- \(s\) was correctly sequenced and appears in the dataset at least twice (possibly as a reverse complement);
- \(s\) is incorrect, it appears in the dataset exactly once, and its Hamming distance is 1 with respect to exactly one correct read in the dataset (or its reverse complement).
Return: A list of all corrections in the form “[old read]->[new read]”. (Each correction must be a single symbol substitution, and you may return the corrections in any order.)
34.1 Sample Dataset
>Rosalind_52
TCATC
>Rosalind_44
TTCAT
>Rosalind_68
TCATC
>Rosalind_28
TGAAA
>Rosalind_95
GAGGA
>Rosalind_66
TTTCA
>Rosalind_33
ATCAA
>Rosalind_21
TTGAT
>Rosalind_18
TTTCC34.2 Sample Output
TTCAT->TTGAT
GAGGA->GATGA
TTTCC->TTTCA34.3 Solution
- Parse the FASTA format input to extract reads.
- Identify correct reads:
- Reads that appear at least twice or appear once but their reverse complement also appears at least once.
- Identify incorrect reads:
- Reads that appear exactly once and do not have their reverse complement in the list of correct reads.
- Correct the errors:
- For each incorrect read, find the correct read that has a Hamming distance of 1 with the incorrect read or its reverse complement.
- Output the corrections.
34.4 Step-by-step Implementation
Here’s the revised Python code to achieve the above steps:
def parse_fasta(fasta_str):
"""
Parses a FASTA formatted string.
"""
sequences = {}
current_label = None
for line in fasta_str.strip().split("\n"):
if line.startswith(">"):
current_label = line[1:].strip()
sequences[current_label] = ""
else:
sequences[current_label] += line.strip()
return sequences
def reverse_complement(dna):
"""
Returns the reverse complement of a DNA string.
"""
complement = {'A': 'T', 'T': 'A', 'C': 'G', 'G': 'C'}
return "".join(complement[base] for base in reversed(dna))
def hamming_distance(s1, s2):
"""
Calculates the Hamming distance between two strings.
"""
return sum(1 for a, b in zip(s1, s2) if a != b)
def find_correct_reads(reads):
"""
Identifies the correct reads in the dataset.
"""
from collections import defaultdict
read_counts = defaultdict(int)
for read in reads:
read_counts[read] += 1
read_counts[reverse_complement(read)] += 1
correct_reads = {read for read, count in read_counts.items() if count > 1}
return correct_reads
def find_corrections(reads, correct_reads):
"""
Identifies corrections needed for the erroneous reads.
"""
corrections = []
for read in reads:
if read not in correct_reads:
for correct_read in correct_reads:
if hamming_distance(read, correct_read) == 1:
corrections.append(f"{read}->{correct_read}")
break
elif hamming_distance(read, reverse_complement(correct_read)) == 1:
corrections.append(f"{read}->{reverse_complement(correct_read)}")
break
return corrections
# Sample Input in FASTA format
fasta_input = """
>Rosalind_52
TCATC
>Rosalind_44
TTCAT
>Rosalind_68
TCATC
>Rosalind_28
TGAAA
>Rosalind_95
GAGGA
>Rosalind_66
TTTCA
>Rosalind_33
ATCAA
>Rosalind_21
TTGAT
>Rosalind_18
TTTCC
"""
# Parsing the input
sequences = parse_fasta(fasta_input)
reads = list(sequences.values())
# Find correct reads
correct_reads = find_correct_reads(reads)
# Find necessary corrections
corrections = find_corrections(reads, correct_reads)
# Output the corrections
for correction in corrections:
print(correction)34.5 Explanation
parse_fasta(fasta_str): Parses the input FASTA formatted string to extract the reads.reverse_complement(dna): Returns the reverse complement of a given DNA string.hamming_distance(s1, s2): Computes the Hamming distance between two strings.find_correct_reads(reads): Identifies reads that are correct (appear at least twice considering both original and reverse complement).find_corrections(reads, correct_reads): Identifies the necessary corrections for erroneous reads by checking each read against the set of correct reads and their reverse complements.
35 Counting Phylogenetic Ancestors
A binary tree is a tree in which each node has degree equal to at most 3. The binary tree will be our main tool in the construction of phylogenies.
A rooted tree is a tree in which one node (the root) is set aside to serve as the pinnacle of the tree. A standard graph theory exercise is to verify that for any two nodes of a tree, exactly one path connects the nodes. In a rooted tree, every node \(v\) will therefore have a single parent, or the unique node \(w\) such that the path from \(v\) to the root contains \({v,w}\). Any other node \(x\) adjacent to \(v\) is called a child of \(v\) because \(v\) must be the parent of \(x\); note that a node may have multiple children. In other words, a rooted tree possesses an ordered hierarchy from the root down to its leaves, and as a result, we may often view a rooted tree with undirected edges as a directed graph in which each edge is oriented from parent to child. We should already be familiar with this idea; it’s how the Rosalind problem tree works!
Even though a binary tree can include nodes having degree 2, an unrooted binary tree is defined more specifically: all internal nodes have degree 3. In turn, a rooted binary tree is such that only the root has degree 2 (all other internal nodes have degree 3).
Given: A positive integer \(n\) (\(3≤n≤10000\)).
Return: The number of internal nodes of any unrooted binary tree having \(n\) leaves.
35.1 Sample Dataset
435.2 Sample Output
235.3 Solution
To solve the problem of finding the number of internal nodes in an unrooted binary tree given \(n\) leaves, let’s delve into some tree properties and the characteristics of unrooted binary trees.
35.4 Key Concepts and Approach
- Tree Properties:
- An unrooted binary tree is a tree where every internal node has exactly three connections (degree 3), and each leaf node has one connection (degree 1).
- Leaves and Internal Nodes Relationship:
- In any tree, if we let \(L\) be the number of leaves and \(I\) be the number of internal nodes, for an unrooted binary tree, there is a specific relationship:
- For every new leaf added to maintain the tree as binary, you essentially add a new internal node to accommodate the structure.
- Mathematical Relationship:
- It is known that for an unrooted binary tree with \(n\) leaves, the number of internal nodes \(I\) is given by: [ I = n - 2 ]
- This is derived from the fact that the total number of nodes in an unrooted binary tree with \(n\) leaves is \(2n - 2\). Out of these, \(n\) are leaves, and the remaining \(n - 2\) are internal nodes.
35.5 Implementation
Given this understanding, the implementation to find the number of internal nodes in an unrooted binary tree with \(n\) leaves is straightforward. Here’s the Python code to accomplish this:
def count_internal_nodes(n):
return n - 2
# Sample Input
n = 4
print(count_internal_nodes(n)) # Output should be 236 k-Mer Composition
For a fixed positive integer \(k\), order all possible k-mers taken from an underlying alphabet lexicographically.
Then the k-mer composition of a string \(s\) can be represented by an array \(A\) for which \(A[m]\) denotes the number of times that the \(m\)th k-mer (with respect to the lexicographic order) appears in \(s\).
Given: A DNA string \(s\) in FASTA format (having length at most 100 kbp).
Return: The 4-mer composition of \(s\).
36.1 Sample Dataset
>Rosalind_6431
CTTCGAAAGTTTGGGCCGAGTCTTACAGTCGGTCTTGAAGCAAAGTAACGAACTCCACGG
CCCTGACTACCGAACCAGTTGTGAGTACTCAACTGGGTGAGAGTGCAGTCCCTATTGAGT
TTCCGAGACTCACCGGGATTTTCGATCCAGCCTCAGTCCAGTCTTGTGGCCAACTCACCA
AATGACGTTGGAATATCCCTGTCTAGCTCACGCAGTACTTAGTAAGAGGTCGCTGCAGCG
GGGCAAGGAGATCGGAAAATGTGCTCTATATGCGACTAAAGCTCCTAACTTACACGTAGA
CTTGCCCGTGTTAAAAACTCGGCTCACATGCTGTCTGCGGCTGGCTGTATACAGTATCTA
CCTAATACCCTTCAGTTCGCCGCACAAAAGCTGGGAGTTACCGCGGAAATCACAG36.2 Sample Output
4 1 4 3 0 1 1 5 1 3 1 2 2 1 2 0 1 1 3 1 2 1 3 1 1 1 1 2 2 5 1 3 0 2 2 1 1 1 1 3 1 0 0 1 5 5 1 5 0 2 0 2 1 2 1 1 1 2 0 1 0 0 1 1 3 2 1 0 3 2 3 0 0 2 0 8 0 0 1 0 2 1 3 0 0 0 1 4 3 2 1 1 3 1 2 1 3 1 2 1 2 1 1 1 2 3 2 1 1 0 1 1 3 2 1 2 6 2 1 1 1 2 3 3 3 2 3 0 3 2 1 1 0 0 1 4 3 0 1 5 0 2 0 1 2 1 3 0 1 2 2 1 1 0 3 0 0 4 5 0 3 0 2 1 1 3 0 3 2 2 1 1 0 2 1 0 2 2 1 2 0 2 2 5 2 2 1 1 2 1 2 2 2 2 1 1 3 4 0 2 1 1 0 1 2 2 1 1 1 5 2 0 3 2 1 1 2 2 3 0 3 0 1 3 1 2 3 0 2 1 2 2 1 2 3 0 1 2 3 1 1 3 1 0 1 1 3 0 2 1 2 2 0 2 1 136.3 Solution
To solve the problem of finding the 4-mer composition of a given DNA string, we need to follow these steps:
- Parse the input FASTA format to extract the DNA sequence.
- Generate all possible 4-mers from the given DNA alphabet.
- Count the occurrences of each 4-mer in the DNA sequence.
- Output the counts in lexicographical order of the 4-mers.
36.4 Detailed Explanation
Parsing the FASTA format: The input DNA sequence is provided in FASTA format. We need to extract the actual DNA string from this format.
Generating all possible 4-mers: A 4-mer is a sequence of 4 nucleotides. Since the DNA alphabet consists of {A, C, G, T}, there are $4^4 = 256) possible 4-mers. We can generate these 4-mers lexicographically (sorted order).
Counting occurrences of each 4-mer: We will slide a window of length 4 acros the DNA sequence and count how many times each 4-mer appears.
Output the results: We output the counts of each 4-mer in the lexicographical order.
36.5 Python Implementation
Here is the complete Python code that implements the above steps:
from itertools import product
def parse_fasta(fasta_str):
sequences = {}
current_label = None
for line in fasta_str.strip().split("\n"):
if line.startswith(">"):
current_label = line[1:].strip()
sequences[current_label] = ""
else:
sequences[current_label] += line.strip()
return sequences
def generate_kmers(k, alphabet='ACGT'):
return [''.join(p) for p in product(alphabet, repeat=k)]
def count_kmers(dna, k):
kmer_counts = {}
for i in range(len(dna) - k + 1):
kmer = dna[i:i+k]
if kmer in kmer_counts:
kmer_counts[kmer] += 1
else:
kmer_counts[kmer] = 1
return kmer_counts
def kmer_composition(dna, k=4):
kmers = generate_kmers(k)
kmer_counts = count_kmers(dna, k)
return [kmer_counts.get(kmer, 0) for kmer in kmers]
# Sample Input in FASTA format
fasta_input = """
>Rosalind_6431
CTTCGAAAGTTTGGGCCGAGTCTTACAGTCGGTCTTGAAGCAAAGTAACGAACTCCACGG
CCCTGACTACCGAACCAGTTGTGAGTACTCAACTGGGTGAGAGTGCAGTCCCTATTGAGT
TTCCGAGACTCACCGGGATTTTCGATCCAGCCTCAGTCCAGTCTTGTGGCCAACTCACCA
AATGACGTTGGAATATCCCTGTCTAGCTCACGCAGTACTTAGTAAGAGGTCGCTGCAGCG
GGGCAAGGAGATCGGAAAATGTGCTCTATATGCGACTAAAGCTCCTAACTTACACGTAGA
CTTGCCCGTGTTAAAAACTCGGCTCACATGCTGTCTGCGGCTGGCTGTATACAGTATCTA
CCTAATACCCTTCAGTTCGCCGCACAAAAGCTGGGAGTTACCGCGGAAATCACAG
"""
# Parsing the input
sequences = parse_fasta(fasta_input)
# There should be only one sequence in the given input
dna_string = list(sequences.values())[0]
# Getting the 4-mer composition
composition = kmer_composition(dna_string, k=4)
# Printing the result
print(" ".join(map(str, composition)))36.6 Explanation of the Code
- parse_fasta(fasta_str): This function parses the input FASTA format string and returns a dictionary of sequences.
- generate_kmers(k, alphabet=‘ACGT’): This function generates all possible k-mers of length
kusing the given alphabet. - count_kmers(dna, k): This function counts the occurrences of each k-mer in the DNA sequence.
- kmer_composition(dna, k=4): This function calculates the k-mer composition by using the previous two functions. It returns a list of counts of each k-mer in lexicographical order.
- The main block: Parses the input, extracts the DNA sequence, computes the 4-mer composition, and prints the results.
37 Speeding Up Motif Finding
A prefix of a length \(n\) string \(s\) is a substring \(s[1:j]\); a suffix of \(s\) is a substring \(s[k:n]\).
The failure array of \(s\) is an array \(P\) of length \(n\) for which \(P[k]\) is the length of the longest substring \(s[j:k]\) that is equal to some prefix \(s[1:k−j+1]\), where \(j\) cannot equal 11 (otherwise, \(P[k]\) would always equal \(k\)). By convention, \(P[1]=0\).
Given: A DNA string \(s\) (of length at most 100 kbp) in FASTA format.
Return: The failure array of \(s\).
37.1 Sample Dataset
>Rosalind_87
CAGCATGGTATCACAGCAGAG37.2 Sample Output
0 0 0 1 2 0 0 0 0 0 0 1 2 1 2 3 4 5 3 0 037.3 Solution
To solve the problem of computing the failure array of a given DNA string in FASTA format, we need to follow these steps:
- Parse the input FASTA format to extract the DNA sequence.
- Compute the failure array using the Knuth-Morris-Prat (KMP) algorithm.
- Output the failure array.
37.4 Detailed Explanation
Parsing the FASTA format: We need to extract the actual DNA string from the provided FASTA format.
Computing the Failure Array: The failure array is computed using the KMP preprocessing algorithm. The failure array
Pat positionkrepresents the length of the longest prefix of the substrings[1:k]that is also a suffix of this substring.
37.5 Python Implementation
Here is the complete Python code that implements the above steps:
def parse_fasta(fasta_str):
"""
Parses a FASTA format string and returns the DNA sequence.
"""
sequences = []
for line in fasta_str.strip().split("\n"):
if not line.startswith(">"):
sequences.append(line.strip())
return "".join(sequences)
def compute_failure_array(s):
"""
Computes the failure array for a given string s using the KMP algorithm.
"""
n = len(s)
P = [0] * n
k = 0
for i in range(1, n):
while k > 0 and s[k] != s[i]:
k = P[k - 1]
if s[k] == s[i]:
k += 1
P[i] = k
return P
# Sample Input in FASTA format
fasta_input = """
>Rosalind_87
CAGCATGGTATCACAGCAGAG
"""
# Parsing the input
dna_string = parse_fasta(fasta_input)
# Computing the failure array
failure_array = compute_failure_array(dna_string)
# Printing the result
print(" ".join(map(str, failure_array)))37.6 Explanation of the Code
- parse_fasta(fasta_str): This function parses the input FASTA format string and returns the concatenated DNA sequence.
- compute_failure_array(s): This function computes the failure array for the string
susing the KMP algorithm.- Initialize an array
Pof lengthnwith zeros. - Iterate through the string
sfrom the second character to the end. - For each character, update the value of
kto the length of the longest prefix which is also a suffix for the substrings[1:i+1]. - Store the value of
kinP[i].
- Initialize an array
- The main block:
- Parse the input FASTA format string to extract the DNA sequence.
- Compute the failure array for the DNA sequence.
- Print the failure array as a space-separated string.
39 Ordering Strings of Varying Length Lexicographically
Say that we have strings \(s=s1s2⋯sm\) and \(t=t1t2⋯tn\) with \(m<n\). Consider the substring \(t′=t[1:m]\). We have two cases:
- If \(s=t′\), then we set \(s<Lext\) because \(s\) is shorter than \(t\) (e.g., \(APPLE<APPLET\)).
- Otherwise, \(s≠t′\). We define \(s<Lext\) if \(s<Lext′\) and define \(s>Lext\) if \(s>Lext′\) (e.g., \(APPLET<LexARTS\) because \(APPL<LexARTS\)).
Given: A permutation of at most 12 symbols defining an ordered alphabet \(A\) and a positive integer \(n\) (\(n≤4\)).
Return: All strings of length at most \(n\) formed from \(A\), ordered lexicographically. (Note: As in “Enumerating k-mers Lexicographically”, alphabet order is based on the order in which the symbols are given.)
39.1 Sample Dataset
D N A
339.2 Sample Output
D
DD
DDD
DDN
DDA
DN
DND
DNN
DNA
DA
DAD
DAN
DAA
N
ND
NDD
NDN
NDA
NN
NND
NNN
NNA
NA
NAD
NAN
NAA
A
AD
ADD
ADN
ADA
AN
AND
ANN
ANA
AA
AAD
AAN
AAA39.3 Solution
To solve the problem of generating all strings of length up to \(n\) formed from an ordered alphabet \(A\), and then ordering them lexicographically based on the given alphabet order, we can use a recursive approach or itertools to generate the permutations. Here is a detailed step-by-step explanation and implementation:
39.4 Steps to Solve the Problem
- Input Parsing:
- Parse the given ordered alphabet \(A\) and the integer \(n\).
- Generate All Possible Strings:
- Use recursion or itertools to generate all strings of length from 1 to \(n\) using the symbols in \(A\).
- Sorting:
- Sort the generated strings based on the custom order provided by \(A\).
39.5 Detailed Explanation
- Generating Combinations:
- For each length \(k\) from 1 to \(n\), generate all possible strings of that length using the symbols in \(A\).
- Custom Sorting:
- Use the order of symbols in \(A\) to sort the generated strings lexicographically.
39.6 Python Implementation
Here is the Python code to achieve the above steps:
import itertools
def parse_input(input_str):
lines = input_str.strip().split("\n")
alphabet = lines[0].split()
n = int(lines[1])
return alphabet, n
def generate_strings(alphabet, n):
all_strings = []
for length in range(1, n + 1):
for combo in itertools.product(alphabet, repeat=length):
all_strings.append("".join(combo))
return all_strings
def custom_sort(strings, alphabet):
order_map = {char: idx for idx, char in enumerate(alphabet)}
return sorted(strings, key=lambda word: [order_map[char] for char in word])
# Sample Input
input_data = """
D N A
3
"""
# Parse input
alphabet, n = parse_input(input_data)
# Generate all strings of length up to n
all_strings = generate_strings(alphabet, n)
# Sort the strings based on the custom lexicographical order
sorted_strings = custom_sort(all_strings, alphabet)
# Print the result
for s in sorted_strings:
print(s)39.7 Explanation of the Code
- parse_input(input_str): This function parses the input string to extract the alphabet and the integer \(n\).
- generate_strings(alphabet, n): This function generates all possible strings of lengths from 1 to \(n\) using itertools.product.
- custom_sort(strings, alphabet): This function sorts the generated strings based on the custom order defined by the alphabet. It uses a mapping of characters to their indices in the given order for sorting.
- Main Execution:
- Parse the input data.
- Generate all possible strings.
- Sort the strings using the custom lexicographical order.
- Print each string in the sorted list.
40 Maximum Matchings and RNA Secondary Structures
The graph theoretical analogue of the quandary stated in the introduction above is that if we have an RNA string s that does not have the same number of occurrences of ‘C’ as ‘G’ and the same number of occurrences of ‘A’ as ‘U’, then the bonding graph of \(s\) cannot possibly posses a perfect matching among its basepair edges. For example, see Figure 1; in fact, most bonding graphs will not contain a perfect matching.
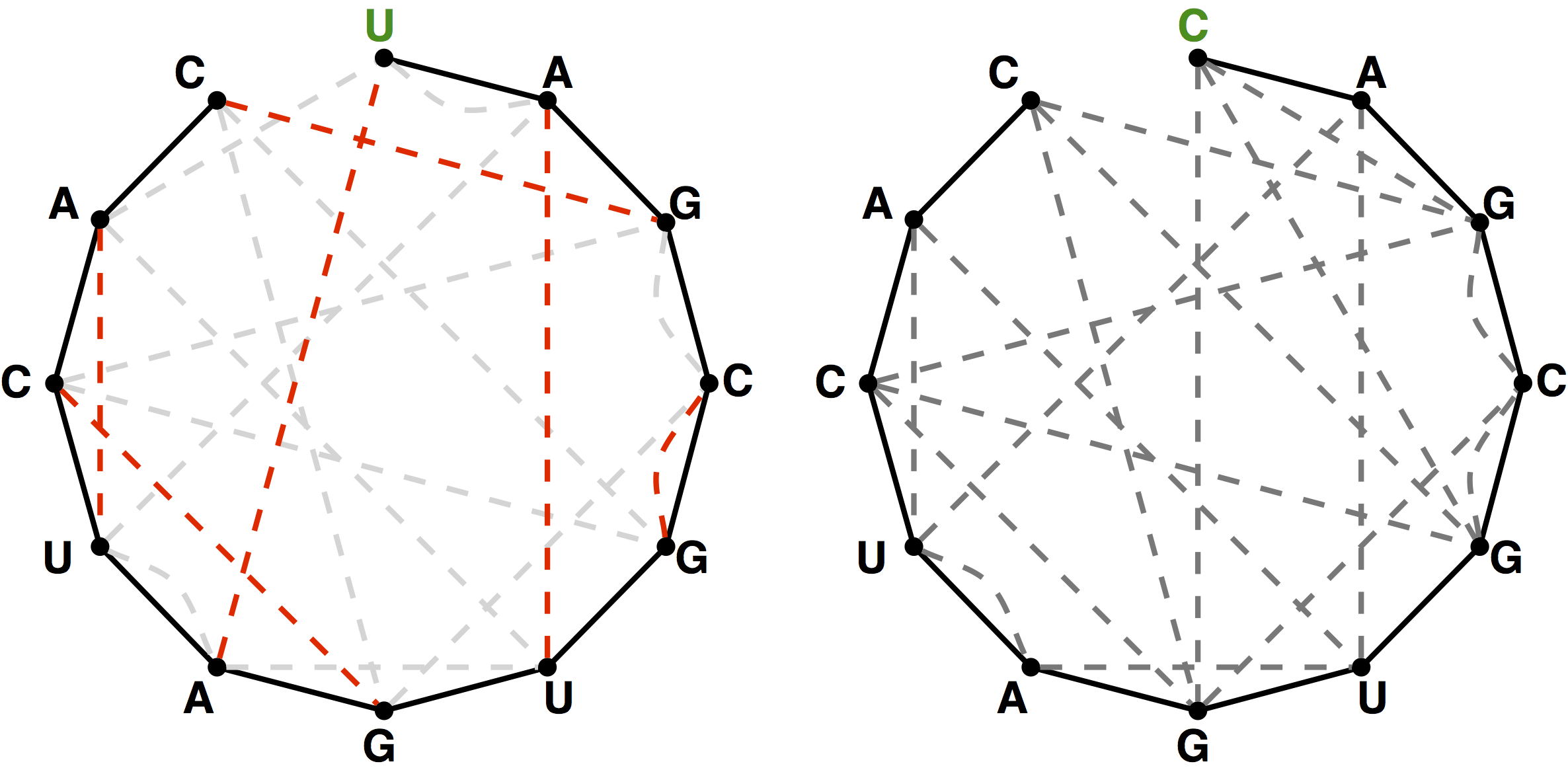{kind=link}
In light of this fact, we define a maximum matching in a graph as a matching containing as many edges as possible.
A maximum matching of basepair edges will correspond to a way of forming as many base pairs as possible in an RNA string.
Given: An RNA string \(s\) of length at most 100.
Return: The total possible number of maximum matchings of basepair edges in the bonding graph of s𝑠.
40.1 Sample Dataset
>Rosalind_92
AUGCUUC40.2 Sample Output
640.3 Solution
- Counting Nucleotides: Count occurrences of each nucleotide.
- Calculating Pairings:
- The number of possible AU pairs is determined by the minimum of A and U.
- Similarly, the number of possible GC pairs is determined by the minimum of G and C.
- Using Factorials: Calculate how many ways to pair these nucleotides.
Let’s correct the implementation:
from math import factorial
def count_nucleotides(sequence):
"""
Count the occurrences of each nucleotide in the RNA sequence.
Args:
sequence (str): The RNA sequence as a string.
Returns:
dict: A dictionary with counts for 'A', 'U', 'G', and 'C'.
"""
counts = {'A': 0, 'U': 0, 'G': 0, 'C': 0}
for nucleotide in sequence:
if nucleotide in counts:
counts[nucleotide] += 1
return counts
def max_matching_pairs(a, b):
"""
Calculate the number of ways to pair 'a' items with 'b' items.
Args:
a (int): Number of items of type A.
b (int): Number of items of type B.
Returns:
int: The number of maximum matching pairs.
"""
return factorial(a) // (factorial(b) * factorial(a - b))
def maximum_matchings(sequence):
"""
Calculate the number of maximum matchings in an RNA sequence.
Args:
sequence (str): The RNA sequence.
Returns:
int: The number of maximum matchings.
"""
# Count nucleotides
counts = count_nucleotides(sequence)
# Get counts for each nucleotide
A = counts['A']
U = counts['U']
G = counts['G']
C = counts['C']
# Calculate the number of possible AU and GC pairings
min_au_pairs = min(A, U)
min_gc_pairs = min(G, C)
# Compute number of ways to form these pairs
au_pairings = max_matching_pairs(A, min_au_pairs) * max_matching_pairs(U, min_au_pairs)
gc_pairings = max_matching_pairs(G, min_gc_pairs) * max_matching_pairs(C, min_gc_pairs)
# Total number of maximum matchings
total_matchings = au_pairings * gc_pairings
return total_matchings
# Example RNA sequence
rna_sequence = "AUGCUUC"
# Compute and print the number of maximum matchings
print(maximum_matchings(rna_sequence))40.4 Explanation of Changes
count_nucleotides(sequence): Counts the number of each nucleotide.max_matching_pairs(a, b): Calculates the number of ways to formbpairs fromaitems using factorials. This function helps in calculating possible pairings for nucleotides.maximum_matchings(sequence): Computes the number of ways to matchAwithUandGwithC, and then multiplies these to get the total number of matchings.
Rosalind 의 서버는 python2.7 로 구현되어 있어 python3 에서 실행한 결과는 옳지 않다고 판단합니다. 따라서 상대적으로 정확도가 떨어지는 python2.7 로 실행하세요.
41 Creating a Distance Matrix
For two strings \(s1\) and \(s2\) of equal length, the p-distance between them, denoted \(dp(s1,s2)\), is the proportion of corresponding symbols that differ between \(s1\) and \(s2\).
For a general distance function \(d\) on \(n\) taxa \(s1,s2,…,sn\) (taxa are often represented by genetic strings), we may encode the distances between pairs of taxa via a distance matrix \(D\) in which \(D_{i,j}=d(s_i,s_j)\).
Given: A collection of \(n\) \((n≤10)\) DNA strings \(s1,…,sn\) of equal length (at most 1 kbp). Strings are given in FASTA format.
Return: The matrix \(D\) corresponding to the p-distance \(d_p\) on the given strings. As always, note that your answer is allowed an absolute error of 0.001.
41.1 Sample Dataset
>Rosalind_9499
TTTCCATTTA
>Rosalind_0942
GATTCATTTC
>Rosalind_6568
TTTCCATTTT
>Rosalind_1833
GTTCCATTTA41.2 Sample Output
0.00000 0.40000 0.10000 0.10000
0.40000 0.00000 0.40000 0.30000
0.10000 0.40000 0.00000 0.20000
0.10000 0.30000 0.20000 0.0000041.3 Solution
To create a distance matrix based on the p-distance for a given set of DNA strings, we will follow these steps:
- Read the Input: Parse the FASTA formatted input to extract DNA strings.
- Calculate p-distance: Compute the p-distance between each pair of DNA strings. The p-distance is defined as the proportion of differing symbols between two strings of equal length.
- Construct the Distance Matrix: Populate the distance matrix with the computed p-distances.
41.4 Detailed Implementation
- Reading the Input: We’ll parse the input to extract the DNA sequences.
- Computing p-distance: For each pair of sequences, we’ll count the differing positions and divide by the total length of the sequences.
- Output the Distance Matrix: Format the matrix with each entry having a precision up to five decimal places.
Here’s the Python code to achieve this:
def read_fasta(fasta_string):
"""
Parses a FASTA formatted string and returns a list of sequences.
"""
sequences = []
current_sequence = []
for line in fasta_string.strip().split('\n'):
if line.startswith('>'):
if current_sequence:
sequences.append(''.join(current_sequence))
current_sequence = []
else:
current_sequence.append(line.strip())
if current_sequence:
sequences.append(''.join(current_sequence))
return sequences
def p_distance(s1, s2):
"""
Computes the p-distance between two DNA sequences of equal length.
"""
assert len(s1) == len(s2), "Sequences must be of equal length."
differences = sum(1 for a, b in zip(s1, s2) if a != b)
return differences / len(s1)
def distance_matrix(sequences):
"""
Computes the distance matrix for a list of sequences based on p-distance.
"""
n = len(sequences)
matrix = [[0] * n for _ in range(n)]
for i in range(n):
for j in range(n):
if i != j:
matrix[i][j] = p_distance(sequences[i], sequences[j])
return matrix
def format_matrix(matrix):
"""
Formats the matrix for output with each entry having five decimal places.
"""
return '\n'.join(' '.join(f"{cell:f}" for cell in row) for row in matrix)
# Sample dataset
fasta_string = """>Rosalind_9499
TTTCCATTTA
>Rosalind_0942
GATTCATTTC
>Rosalind_6568
TTTCCATTTT
>Rosalind_1833
GTTCCATTTA"""
# Reading sequences from the sample dataset
sequences = read_fasta(fasta_string)
# Calculating the distance matrix
dist_matrix = distance_matrix(sequences)
# Formatting and printing the distance matrix
formatted_matrix = format_matrix(dist_matrix)
print(formatted_matrix)41.5 Explanation
- Reading FASTA Data: The
read_fastafunction processes the input FASTA data and extracts the sequences. - Calculating p-distance: The
p_distancefunction computes the proportion of differing symbols between two sequences. - Constructing Distance Matrix: The
distance_matrixfunction creates a matrix where each entry $(i, j)) contains the p-distance between sequences $i) and $j). - Formatting the Output: The
format_matrixfunction ensures that each entry in the matrix is printed with five decimal places for precision.
42 Reversal Distance
A reversal of a permutation creates a new permutation by inverting some interval of the permutation; \((5,2,3,1,4)\), \((5,3,4,1,2)\), and \((4,1,2,3,5)\), are all reversals of \((5,3,2,1,4)\). The reversal distance between two permutations π𝜋 and σ𝜎, written \(d_rev(π,σ)\), is the minimum number of reversals required to transform \(π\) into \(σ\) (this assumes that \(π\) and \(σ\) have the same length).
Given: A collection of at most 5 pairs of permutations, all of which have length 10.
Return: The reversal distance between each permutation pair.
42.1 Sample Dataset
1 2 3 4 5 6 7 8 9 10
3 1 5 2 7 4 9 6 10 8
3 10 8 2 5 4 7 1 6 9
5 2 3 1 7 4 10 8 6 9
8 6 7 9 4 1 3 10 2 5
8 2 7 6 9 1 5 3 10 4
3 9 10 4 1 8 6 7 5 2
2 9 8 5 1 7 3 4 6 10
1 2 3 4 5 6 7 8 9 10
1 2 3 4 5 6 7 8 9 1042.2 Sample Output
9 4 5 7 042.3 Solution
import collections
def get_all_permutations(s):
for i in range(len(s)):
for j in range(i + 2, len(s) + 1):
yield s[:i] + s[i:j][::-1] + s[j:]
def get_reversal_distance(p1, p2):
if p1 == p2:
return 0
target = tuple(p2)
fromfirst = {tuple(p1): 0}
q = collections.deque([p1])
while q:
s = q.popleft()
c = fromfirst[s]
for j in get_all_permutations(s):
if j == target:
return c + 1
if j not in fromfirst:
fromfirst[j] = c + 1
if c != 4:
q.append(j)
fromsecond = {tuple(p2): 0}
target = tuple(p1)
q = collections.deque([p2])
answer = 100000
while q:
s = q.popleft()
c = fromsecond[s]
if c == 4:
break
for j in get_all_permutations(s):
if j == target:
return c + 1
if j not in fromsecond:
fromsecond[j] = c + 1
if c != 3:
q.append(j)
if j in fromfirst:
answer = min(answer, fromfirst[j] + fromsecond[j])
return answer
input_data = """
1 2 3 4 5 6 7 8 9 10
3 1 5 2 7 4 9 6 10 8
3 10 8 2 5 4 7 1 6 9
5 2 3 1 7 4 10 8 6 9
8 6 7 9 4 1 3 10 2 5
8 2 7 6 9 1 5 3 10 4
3 9 10 4 1 8 6 7 5 2
2 9 8 5 1 7 3 4 6 10
1 2 3 4 5 6 7 8 9 10
1 2 3 4 5 6 7 8 9 10
"""
# Proces input data
dataset = list(map(str.strip, input_data.strip().split('\n')))
distances = []
for i in range(0, len(dataset), 3):
s = tuple(map(int, dataset[i].split()))
t = tuple(map(int, dataset[i + 1].split()))
distances.append(get_reversal_distance(t, s))
print(' '.join(map(str, distances)))42.4 Key Changes and Additions
- Conversion to Python 3:
- Changed
xrangetorange. - Changed
printstatement toprint()function.
- Changed
- Input Handling:
- Replaced file reading with a direct
input_datastring for simplicity and demonstration purposes. - Processed the
input_datastring to split it into individual lines and then handled them similarly to how they would be read from a file.
- Replaced file reading with a direct
43 Counting Subsets
A set is the mathematical term for a loose collection of objects, called elements. Examples of sets include \({the moon, the sun, Wilford Brimley}\) and \(R\), the set containing all real numbers. We even have the empty set, represented by \(∅\) or \({}\), which contains no elements at all. Two sets are equal when they contain the same elements. In other words, in contrast to permutations, the ordering of the elements of a set is unimportant (e.g., \({the moon, the sun, Wilford Brimley}\) is equivalent to \({Wilford Brimley, the moon, the sun}\)). Sets are not allowed to contain duplicate elements, so that \({Wilford Brimley, the sun, the sun}\) is not a set. We have already used sets of 2 elements to represent edges from a graph.
A set \(A\) is a subset of \(B\)𝐵 if every element of \(A\) is also an element of B𝐵, and we write \(A⊆B\). For example, \({the sun, the moon}⊆{the sun, the moon, Wilford Brimley}\), and \(∅\) is a subset of every set (including itself!).
As illustrated in the biological introduction, we can use subsets to represent the collection of taxa possessing a character. However, the number of applications is endless; for example, an event in probability can now be defined as a subset of the set containing all possible outcomes.
Our first question is to count the total number of possible subsets of a given set.
Given: A positive integer \(n\) (\(n≤1000\)).
Return: The total number of subsets of \({1,2,…,n}\) modulo 1,000,000.
43.1 Sample Dataset
343.2 Sample Output
843.3 Solution
To solve the problem of counting the total number of subsets of the set \({1, 2, \ldots, n}\) modulo 1,000,000, we need to understand a few key points:
- Subsets of a Set:
- For any set of size \(n\), the number of possible subsets is \(2^n\). This includes the empty set and the set itself.
- Modulo Operation:
- Since \(n\) can be as large as 1000, \(2^n\) can be a very large number. To manage this, we will compute the result modulo 1,000,000.
43.4 Steps to Solution
- Calculate \(2^n \mod 1,000,000\):
- We will use modular exponentiation to compute this efficiently. Direct computation of \(2^n\) for large \(n\) is impractical due to the size of the number.
- Modular Exponentiation:
- This technique allows us to compute \((base^{exp}) \mod mod\) efficiently using an iterative or recursive approach that reduces the number of multiplications required.
Here’s the Python code to solve the problem:
def modular_exponentiation(base, exp, mod):
result = 1
base = base % mod
while exp > 0:
if (exp % 2) == 1: # If exp is odd, multiply base with result
result = (result * base) % mod
exp = exp >> 1 # exp = exp // 2
base = (base * base) % mod # Change base to base^2
return result
def count_subsets(n):
mod = 1000000
return modular_exponentiation(2, n, mod)
# Sample input
n = 3
print(count_subsets(n)) # Output should be 843.5 Explanation of the Code
- Function
modular_exponentiation:- Inputs:
base(2),exp(n), andmod(1,000,000). - Process: This function uses an efficient loop to compute the exponentiation modulo
mod. By squaring the base and halving the exponent iteratively, it ensures that we keep the numbers manageable and perform fewer multiplications.
- Inputs:
- Function
count_subsets:- This function simply calls
modular_exponentiationwithbase2,expn, andmod1,000,000.
- This function simply calls
- Main Execution:
- The sample input
n = 3is used to demonstrate the function, which should output8as expected. - The script can also read from standard input for actual use cases.
- The sample input
44 Matching Random Motifs
Our aim in this problem is to determine the probability with which a given motif (a known promoter, say) occurs in a randomly constructed genome. Unfortunately, finding this probability is tricky; instead of forming a long genome, we will form a large collection of smaller random strings having the same length as the motif; these smaller strings represent the genome’s substrings, which we can then test against our motif.
Given a probabilistic event \(A\), the complement of \(A\) is the collection \(A^c\) of outcomes not belonging to \(A\). Because \(A^c\) takes place precisely when \(A\) does not, we may also call \(A^c\) “not \(A\).”
For a simple example, if \(A\) is the event that a rolled die is 2 or 4, then \(Pr(A)= \frac {1}{3}\). \(A^c\) is the event that the die is 1, 3, 5, or 6, and \(Pr(A^c)= \frac{2}{3}\). In general, for any event we will have the identity that \(Pr(A)+Pr(A^c)=1\).
Given: A positive integer \(N≤100000\), a number \(x\) between 0 and 1, and a DNA string \(s\) of length at most 10 bp.
Return: The probability that if \(N\) random DNA strings having the same length as \(s\) are constructed with GC-content \(x\) (see “Introduction to Random Strings”), then at least one of the strings equals s𝑠. We allow for the same random string to be created more than once.
44.1 Sample Dataset
90000 0.6
ATAGCCGA44.2 Sample Output
0.68944.3 Solution
To solve the problem of calculating the probability that at least one out of \(N\)random DNA strings matches a given DNA string \(s\), we can follow these steps:
- Calculate the Probability of Matching a Single String:
- The probability of a specific base in \(s\)matching a random base depends on the GC-content \(x\).
- For GC-content \(x\), the probabilities are:
- Probability of ‘G’ or ‘C’: \(\frac{x}{2}\)
- Probability of ‘A’ or ‘T’: \(\frac{1 - x}{2}\)
- Compute the Probability of the Entire String Matching:
- The probability that a random DNA string of the same length as \(s\)matches \(s\)exactly is the product of the probabilities for each individual base.
- Calculate the Complementary Probability:
- Compute the probability that a single random DNA string does NOT match \(s\).
- Using this, compute the probability that all \(N\)random strings do NOT match \(s\).
- Compute the Final Probability:
- The probability that at least one out of \(N\)random DNA strings matches \(s\)is the complement of the probability that none of them matches \(s\).
Let’s go through the implementation of this step-by-step:
44.4 Implementation
def calculate_probability(N, x, s):
# Step 1: Calculate the probability of matching a single base
prob_match = 1.0
for base in s:
if base in 'GC':
prob_match *= x / 2
else: # base in 'AT'
prob_match *= (1 - x) / 2
# Step 2: Compute the probability of the entire string matching
# This is already computed as prob_match
# Step 3: Calculate the complementary probability
prob_not_match = 1 - prob_match
# Step 4: Compute the final probability
prob_all_not_match = prob_not_match ** N
prob_at_least_one_match = 1 - prob_all_not_match
return prob_at_least_one_match
# Sample input
N = 90000
x = 0.6
s = "ATAGCCGA"
# Calculate and print the probability
result = calculate_probability(N, x, s)
print(f"{result:f}")44.5 Explanation of the Code
- Probability Calculation for Each Base:
- For each base in the string \(s\), the probability of it being either ‘G’ or ‘C’ is \(\frac{x}{2}\) and for ‘A’ or ‘T’ is \(\frac{1 - x}{2}\).
- Computing Probability for the Entire String:
- Multiply the probabilities of each base matching to get the probability of the entire string matching a random string of the same length.
- Complementary Probability:
- Compute the probability that a single random DNA string does NOT match \(s\).
- Raise this probability to the power \(N\) to get the probability that none of the \(N\) strings match \(s\).
- Final Probability:
- Subtract the complementary probability from 1 to get the probability that at least one out of \(N\) random strings matches \(s\).
45 Introduction to Alternative Splicing
In “Counting Subsets”, we saw that the total number of subsets of a set \(S\) containing \(n\) elements is equal to \(2^n\).
However, if we intend to count the total number of subsets of \(S\) having a fixed size \(k\), then we use the combination statistic \(C(n,k)\) also written \(\binom{n}k\).
Given: Positive integers n and \(m\) with \(0≤m≤n≤20000\).
Return: The sum of combinations \(C(n,k)\) for all k satisfying \(m≤k≤n\), modulo 1,000,000. In shorthand, \(\sum^n_{k=m} \binom{n+1}k\).
45.1 Sample Dataset
6 345.2 Sample Output
4245.3 Solution
Here’s the refactored code that performs the required calculation using Python’s built-in math module while maintaining readability and simplicity.
import math
def sum_of_combinations(n, m, MOD):
total = 0
for k in range(m, n + 1):
# Calculate C(n, k) using math.factorial
comb = math.factorial(n) // (math.factorial(k) * math.factorial(n - k))
total = (total + comb) % MOD
return total
# Sample input
sample_input = "6 3"
n, m = map(int, sample_input.strip().split())
# Define the modulo
MOD = 1000000
# Calculate the sum of combinations
result = sum_of_combinations(n, m, MOD)
# Print the results
print(result)45.4 Explanation
- Function Definition:
- Main Logic:
- Iterate through all \(k\)from \(m\)to \(n\).
- Calculate \(C(n, k)\) using the factorial function.
- Sum the results, taking modulo \(1000000\)at each step to handle large numbers efficiently.
- Sample Input Handling:
- Parse the sample input to get the values of \(n\)and \(m\).
- Define the modulo value \(MOD = 1000000\).
- Calculate the result using the
sum_of_combinationsfunction and print the results.
46 Edit Distance
Given two strings \(s\) and \(t\) (of possibly different lengths), the edit distance \(d_E(s,t)\) is the minimum number of edit operations needed to transform \(s\) into \(t\), where an edit operation is defined as the substitution, insertion, or deletion of a single symbol.
The latter two operations incorporate the case in which a contiguous interval is inserted into or deleted from a string; such an interval is called a gap. For the purposes of this problem, the insertion or deletion of a gap of length \(k\) still counts as \(k\) distinct edit operations.
Given: Two protein strings \(s\) and \(t\) in FASTA format (each of length at most 1000 aa).
Return: The edit distance \(d_E(s,t)\).
46.1 Sample Dataset
>Rosalind_39
PLEASANTLY
>Rosalind_11
MEANLY46.2 Sample Output
547 Expected Number of Restriction Sites
Say that you place a number of bets on your favorite sports teams. If their chances of winning are 0.3, 0.8, and 0.6, then you should expect on average to win \(0.3 + 0.8 + 0.6 = 1.7\) of your bets (of course, you can never win exactly 1.7!)
More generally, if we have a collection of events \(A1,A2,…,An\) then the expected number of events occurring is \(Pr(A1)+Pr(A2)+⋯+Pr(An)\) (consult the note following the problem for a precise explanation of this fact). In this problem, we extend the idea of finding an expected number of events to finding the expected number of times that a given string occurs as a substring of a random string.
Given: A positive integer \(n\) (\(n≤1,000,000\)), a DNA string \(s\) of even length at most 10, and an array \(A\) of length at most 20, containing numbers between 0 and 1.
Return: An array \(B\) having the same length as \(A\) in which \(B[i]\) represents the expected number of times that \(s\) will appear as a substring of a random DNA string \(t\) of length \(n\), where \(t\) is formed with GC-content \(A[i]\) (see “Introduction to Random Strings”).
47.1 Sample Dataset
10
AG
0.25 0.5 0.7547.2 Sample Output
0.422 0.563 0.42247.3 Solution
To solve this problem, we need to compute the expected number of times a given DNA string \(s\)will appear as a substring in a random DNA string \(t\)of length \(n\). The DNA string \(t\)is generated with varying GC-content values given in an array \(A\).
47.4 Approach
- Calculate Probabilities:
- The probability of each nucleotide (A, T, C, G) depends on the GC-content.
- For a given GC-content \(gc\):
- Probability of G or C: \(\frac{gc}{2}\)
- Probability of A or T: \(\frac{1 - gc}{2}\)
- Compute the Probability of \(s\)as a Substring:
- For each GC-content value \(A[i]\), compute the probability that \(s\)appears at any specific position in \(t\).
- Sum the probabilities for all possible starting positions of \(s\)in \(t\)(from 0 to \(n - \text{len}(s)\)).
- Expected Value:
- Multiply the single-position probability by the number of possible starting positions to get the expected number of times \(s\)appears in \(t\).
47.5 Detailed Steps and Code
- Reading Input:
- Read \(n\), the DNA string \(s\), and the array \(A\).
- Probability Calculation:
- For each GC-content value in \(A\), compute the probability of \(s\).
- Output:
- Print the expected values for each GC-content in \(A\).
Here’s the Python code to achieve this:
def expected_restriction_sites(n, s, A):
len_s = len(s)
B = []
for gc_content in A:
p_gc = gc_content / 2
p_at = (1 - gc_content) / 2
prob_s = 1.0
for nucleotide in s:
if nucleotide in 'GC':
prob_s *= p_gc
else:
prob_s *= p_at
expected_count = prob_s * (n - len_s + 1)
B.append(expected_count)
return B
# Sample input
text = """
10
AG
0.25 0.5 0.75"""
n = int(text.strip().split("\n")[0])
s = text.strip().split("\n")[1]
A = [float(x) for x in text.strip().split("\n")[2].split()]
result = expected_restriction_sites(n, s, A)
print(" ".join(f"{x:f}" for x in result))47.6 Explanation
- Reading Input:
nis the length of the random DNA string.sis the DNA substring we are looking for.Ais an array of GC-content values.
- Probability Calculation:
- For each GC-content \(gc\), compute the probability
prob_sthat the substring \(s\) will match exactly at a given position. - Use the formula: \[ \text{prob\_s} = \prod_{i=1}^{\text{len}(s)} \text{probability of } s[i] \]
- For each GC-content \(gc\), compute the probability
- Expected Value:
- Multiply
prob_sby the number of possible starting positions in the string \(t\)(which is \(n - \text{len}(s) + 1\)).
- Multiply
- Output:
- Print the expected counts, formatted to three decimal places.
48 Motzkin Numbers and RNA Secondary Structures
Similarly to our definition of the Catalan numbers, the \(n\)-th Motzkin number \(mn\) counts the number of ways to form a (not necessarily perfect) noncrossing matching in the complete graph \(Kn\) containing \(n\) nodes.
How should we compute the Motzkin numbers? As with Catalan numbers, we will take \(m0=m1=1\). To calculate \(mn\) in general, assume that the nodes of \(Kn\) are labeled around the outside of a circle with the integers between 1 and \(n\), and consider node 1, which may or may not be involved in a matching. If node 1 is not involved in a matching, then there are \(m_{n−1}\) ways of matching the remaining \(n−1\) nodes. If node 1 is involved in a matching, then say it is matched to node \(k\): this leaves \(k−2\) nodes on one side of edge \({1,k}\) and \(n−k\) nodes on the other side; as with the Catalan numbers, no edge can connect the two sides, which gives us \(m_{k−2}⋅m_{n−k}\) ways of matching the remaining edges. Allowing \(k\) to vary between 22 and \(n\) yields the following recurrence relation for the Motzkin numbers: \(m_n=m_{n−1} + \sum ^n_{k=2} m_k−2⋅m_{n−k}\).
To count all possible secondary structures of a given RNA string that do not contain pseudoknots, we need to modify the Motzkin recurrence so that it counts only matchings of basepair edges in the bonding graph corresponding to the RNA string.
Given: An RNA string \(s\) of length at most 300 bp.
Return: The total number of noncrossing matchings of basepair edges in the bonding graph of \(s\), modulo 1,000,000.
48.1 Sample Dataset
>Rosalind_57
AUAU48.2 Sample Output
748.3 Solution
We want to count the number of ways to form noncrossing matchings of basepair edges in an RNA string. RNA strings can form base pairs between A and U or C and G.
- Initialization:
- Input: An RNA string
rnaof lengthn. - DP Table: We create a 2D list
dpof size(n+1) x (n+1)initialized to zero.dp[i][j]will store the number of noncrossing matchings in the substring from indexitoj-1of the RNA string. - Base Case:
dp[i][i] = 1because a single nucleotide can only be matched with itself.dp[i][i+1] = 1because a pair of adjacent nucleotides can either be unmatched or form one valid base pair.
- Input: An RNA string
- Filling the DP Table:
- We iterate over all possible substring lengths starting from 2 up to
n. - For each substring of length
lengthstarting at indexiand ending at indexj-1:- We start by assuming the first nucleotide
rna[i]is not paired, so the count of valid matchings is initiallydp[i+1][j]. - Then we check all possible positions
kwhererna[i]can form a valid base pair (i.e.,rna[i]withrna[k]). If they form a valid base pair:- We add the number of ways to match the left part (
dp[i+1][k]) and the right part (dp[k+1][j]).
- We add the number of ways to match the left part (
- Sum the results and take modulo $10^6) to avoid large numbers.
- We start by assuming the first nucleotide
- We iterate over all possible substring lengths starting from 2 up to
- Result:
- The result for the entire RNA string is stored in
dp[0][n].
- The result for the entire RNA string is stored in
48.4 Code Explanation
Here’s the code again with comments to explain each part:
def count_noncrossing_matchings(rna):
n = len(rna) # Length of the RNA string
MOD = 1000000 # Modulo value to avoid large numbers
# Initialize a dp table with all zeros
dp = [[0] * (n + 1) for _ in range(n + 1)]
# Base case: Single nucleotides and empty string
for i in range(n + 1):
dp[i][i] = 1 # A single nucleotide matches with itself
if i < n:
dp[i][i + 1] = 1 # Two adjacent nucleotides can be unmatched or paired
# Fill the dp table for all substring lengths
for length in range(2, n + 1): # Length of the substring
for i in range(n - length + 1):
j = i + length
dp[i][j] = dp[i + 1][j] # Case when the first nucleotide is unpaired
for k in range(i + 1, j):
# Check if rna[i] and rna[k] can form a valid base pair
if (rna[i] == 'A' and rna[k] == 'U') or (rna[i] == 'U' and rna[k] == 'A') or \
(rna[i] == 'C' and rna[k] == 'G') or (rna[i] == 'G' and rna[k] == 'C'):
dp[i][j] += dp[i + 1][k] * dp[k + 1][j]
dp[i][j] %= MOD # Take modulo to avoid large numbers
# The result for the entire string
return dp[0][n]
# Sample input
rna_string = "AUAU"
result = count_noncrossing_matchings(rna_string)
print(result) # Output: 748.5 Key Points
- Base Cases: Handle single and adjacent nucleotides.
- Dynamic Programming: Use a table to store results of subproblems to build up the solution for the entire string.
- Modulo Operation: Keep results manageable by taking modulo $10^6).
This approach efficiently calculates the number of noncrossing matchings for the given RNA string.
49 Distances in Trees
Newick format is a way of representing trees even more concisely than using an adjacency list, especially when dealing with trees whose internal nodes have not been labeled.
First, consider the case of a rooted tree \(T\). A collection of leaves \(v1,v2,…,vn\) of \(T\) are neighbors if they are all adjacent to some internal node \(u\). Newick format for \(T\) is obtained by iterating the following key step: delete all the edges \({vi,u}\) from \(T\) and label \(u\) with \((v1,v2,…,vn)u\). This proces is repeated all the way to the root, at which point a semicolon signals the end of the tree.
A number of variations of Newick format exist. First, if a node is not labeled in \(T\), then we simply leave blank the space occupied by the node. In the key step, we can write \((v1,v2,…,vn)\) in place of \((v1,v2,…,vn)u\) if the \(v_i\) are labeled; if none of the nodes are labeled, we can write \((,,…,)\).
A second variation of Newick format occurs when \(T\) is unrooted, in which case we simply select any internal node to serve as the root of \(T\). A particularly peculiar case of Newick format arises when we choose a leaf to serve as the root.
Note that there will be a large number of different ways to represent \(T\) in Newick format.
Given: A collection of \(n\) trees (\(n≤40\)) in Newick format, with each tree containing at most 200 nodes; each tree \(Tk\) is followed by a pair of nodes \(xk\) and \(yk\) in \(Tk\).
Return: A collection of \(n\) positive integers, for which the \(k\)th integer represents the distance between \(xk\) and \(yk\) in \(Tk\).
49.1 Sample Dataset
(cat)dog;
dog cat
(dog,cat);
dog cat49.2 Sample Output
1 249.3 Solution
def dis_tree(T, x, y):
# Find the indices of x and y in the Newick string T
x_index = T.find(x)
y_index = T.find(y)
# Extract the relevant substring between the indices of x and y
sub_tree = [i for i in T[min(x_index, y_index):max(x_index, y_index)] if i in [')', '(', ',']]
# Convert the list of characters to a string
bracket = ''.join(sub_tree)
# Remove empty pairs of parentheses
while '(,)' in bracket:
bracket = bracket.replace('(,)', '')
# Determine the number of steps based on the type of brackets remaining
if bracket.count('(') == len(bracket) or bracket.count(')') == len(bracket):
return len(bracket)
elif bracket.count(',') == len(bracket):
return 2
else:
return bracket.count(')') + bracket.count('(') + 2
def process_input(input_data):
# Parse the input data into a list of tree and node pairs
tree_data = [line.strip().replace(";", "") for line in input_data.strip().split("\n") if line.strip()]
results = []
# Iterate through the parsed data to proces each tree and node pair
for i in range(0, len(tree_data), 2):
T = tree_data[i]
x, y = tree_data[i+1].split(' ')
results.append(dis_tree(T, x, y))
return results
# Sample input
input_data = """
(cat)dog;
dog cat
(dog,cat);
dog cat
"""
# Proces the input and print the results
output_data = process_input(input_data)
print(" ".join(map(str, output_data))) # Output should be: 1 249.4 Explanation
- The
dis_treefunction computes the distance between nodesxandyin the given Newick stringT. - The
process_inputfunction processes the input string, extracts the tree and node pairs, and computes the distances using thedis_treefunction. - Finally, the results are printed in the required format.
50 Interleaving Two Motifs
A string \(s\) is a supersequence of another string \(t\) if \(s\) contains \(t\) as a subsequence.
A common supersequence of strings \(s\) and \(t\) is a string that serves as a supersequence of both \(s\) and \(t\). For example, “GACCTAGGAACTC” serves as a common supersequence of “ACGTC” and “ATAT”. A shortest common supersequence of \(s\) and \(t\) is a supersequence for which there does not exist a shorter common supersequence. Continuing our example, “ACGTACT” is a shortest common supersequence of “ACGTC” and “ATAT”.
Given: Two DNA strings \(s\) and \(t\).
Return: A shortest common supersequence of \(s\) and \(t\). If multiple solutions exist, you may output any one.
50.1 Sample Dataset
ATCTGAT
TGCATA50.2 Sample Output
ATGCATGAT50.3 Solution
To solve the problem of finding the shortest common supersequence (SCS) of two DNA strings $ s$ and $ t$, we can use a dynamic programming approach. The idea is similar to finding the longest common subsequence (LCS), but with a few modifications to ensure that we construct the SCS.
50.4 Steps to Solve the Problem
- Define the Dynamic Programming Table:
- Let \(dp[i][j]\) represent the length of the SCS of the substrings \(s[0:i]\) and \(t[0:j]\).
- Initialize the Table:
- For \(dp[i][0]\), the SCS is simply the prefix of \(s\) of length \(i\), so \(dp[i][0] = i\).
- For \(dp[0][j]\), the SCS is simply the prefix of \(t\) of length \(j\), so \(dp[0][j] = j\).
- Fill the DP Table:
- If \(s[i-1] == t[j-1]\), then \(dp[i][j] = dp[i-1][j-1] + 1\) because the characters match and they contribute once to the SCS.
- Otherwise, \(dp[i][j] = \min(dp[i-1][j], dp[i][j-1]) + 1\), meaning we take the shorter SCS by either adding the current character of \(s\) or \(t\).
- Construct the SCS:
- Use the DP table to backtrack and construct the SCS by starting from \(dp[len(s)][len(t)]\).
50.5 Code Implementation
Here’s the code to solve the problem:
def shortest_common_supersequence(s, t):
m, n = len(s), len(t)
# Initialize the DP table
dp = [[0] * (n + 1) for _ in range(m + 1)]
# Fill the base cases
for i in range(1, m + 1):
dp[i][0] = i
for j in range(1, n + 1):
dp[0][j] = j
# Fill the DP table
for i in range(1, m + 1):
for j in range(1, n + 1):
if s[i - 1] == t[j - 1]:
dp[i][j] = dp[i - 1][j - 1] + 1
else:
dp[i][j] = min(dp[i - 1][j], dp[i][j - 1]) + 1
# Backtrack to find the SCS
i, j = m, n
scs = []
while i > 0 and j > 0:
if s[i - 1] == t[j - 1]:
scs.append(s[i - 1])
i -= 1
j -= 1
elif dp[i - 1][j] < dp[i][j - 1]:
scs.append(s[i - 1])
i -= 1
else:
scs.append(t[j - 1])
j -= 1
while i > 0:
scs.append(s[i - 1])
i -= 1
while j > 0:
scs.append(t[j - 1])
j -= 1
return ''.join(reversed(scs))
# Sample input
s = "ATCTGAT"
t = "TGCATA"
# Calculate and print the shortest common supersequence
print(shortest_common_supersequence(s, t)) # Output should be a valid SCS like "ATGCATGAT"51 Introduction to Set Operations
If \(A\) and \(B\) are sets, then their union \(A∪B\) is the set comprising any elements in either \(A\) or \(B\); their intersection \(A∩B\) is the set of elements in both \(A\) and \(B\); and their set difference \(A−B\) is the set of elements in \(A\) but not in \(B\).
Furthermore, if \(A\) is a subset of another set \(U\), then the set complement of \(A\) with respect to \(U\) is defined as the set \(A^c=U−A\). See the Sample sections below for examples.
Given: A positive integer \(n\) (\(n≤20,000\)) and two subsets \(A\) and \(B\) of \({1,2,…,n}\).
Return: Six sets: \(A∪B\), \(A∩B\), \(A−B\), \(B−A\), \(A^c\), and \(B^c\) (where set complements are taken with respect to \({1,2,…,n}\).
51.1 Sample Dataset
10
{1, 2, 3, 4, 5}
{2, 8, 5, 10}51.2 Sample Output
{1, 2, 3, 4, 5, 8, 10}
{2, 5}
{1, 3, 4}
{8, 10}
{8, 9, 10, 6, 7}
{1, 3, 4, 6, 7, 9}51.3 Solution
To solve this problem involving set operations, we need to perform union, intersection, set difference, and complement operations on two given sets \(A\) and \(B\), with respect to a universal set \(U\) which contains all integers from 1 to \(n\). Here’s how we can approach this step-by-step:
51.4 Steps
- Read Input:
- The first line contains the integer \(n\), the size of the universal set.
- The second line contains set \(A\).
- The third line contains set \(B\).
- Parse Sets:
- Extract the elements of sets \(A\) and \(B\) from the input strings.
- Define Universal Set \(U\):
- This is simply the set of all integers from 1 to \(n\).
- Perform Set Operations:
- Union \(A \cup B\): Elements in either \(A\) or \(B\).
- Intersection \(A \cap B\): Elements common to both \(A\) and \(B\).
- Difference \(A - B\): Elements in \(A\) but not in \(B\).
- Difference \(B - A\): Elements in \(B\) but not in \(A\).
- Complement \(A^c\): Elements in \(U\) but not in \(A\).
- Complement \(B^c\): Elements in \(U\) but not in \(B\).
Here’s the Python code to implement the above logic:
def set_operations(n, A, B):
U = set(range(1, n+1))
A = set(A)
B = set(B)
union = A | B
intersection = A & B
difference_A_B = A - B
difference_B_A = B - A
complement_A = U - A
complement_B = U - B
return union, intersection, difference_A_B, difference_B_A, complement_A, complement_B
# Sample input
n = 10
A = {1, 2, 3, 4, 5}
B = {2, 8, 5, 10}
# Get the results
results = set_operations(n, A, B)
# Print the results
for result in results:
print(result)51.5 Explanation
- Union (\(A \cup B\)): Combines all unique elements from both sets.
- Intersection (\(A \cap B\)): Selects only the elements that are present in both sets.
- Difference (\(A - B\)): Selects elements that are in \(A\) but not in \(B\).
- Difference (\(B - A\)): Selects elements that are in \(B\) but not in \(A\).
- Complement (\(A^c\)): Selects elements that are in the universal set \(U\) but not in \(A\).
- Complement (\(B^c\)): Selects elements that are in the universal set \(U\) but not in \(B\).
52 Sorting by Reversals
A reversal of a permutation can be encoded by the two indices at the endpoints of the interval that it inverts; for example, the reversal that transforms \((4,1,2,6,3,5)\) into \((4,1,3,6,2,5)\) is encoded by \([3,5]\).
A collection of reversals sorts \(π\) into \(γ\) if the collection contains \(d_{rev}(π,γ)\) reversals, which when successively applied to \(π\) yield \(γ\).
Given: Two permutations π𝜋 and \(γ\), each of length 10.
Return: The reversal distance \(d_{rev}(π,γ)\), followed by a collection of reversals sorting \(π\) into \(γ\). If multiple collections of such reversals exist, you may return any one.
52.1 Sample Dataset
1 2 3 4 5 6 7 8 9 10
1 8 9 3 2 7 6 5 4 1052.2 Sample Output
2
4 9
2 552.3 Solution
To solve the problem of sorting by reversals, we need to find the minimum number of reversals needed to transform one permutation \(\pi\) into another permutation \(\gamma\), as well as the specific reversals that accomplish this transformation.
52.4 Key Concepts
- Reversal: A reversal is an operation that takes a segment of the permutation and reverses the order of the elements within that segment.
- Reversal Distance: The reversal distance between two permutations is the minimum number of reversals needed to transform one permutation into another.
52.5 Approach
- Graph Representation:
- Represent the permutations as nodes in a graph, where each node is a permutation and edges represent a single reversal operation transforming one permutation into another.
- Breadth-First Search (BFS):
- Use BFS to find the shortest path from the starting permutation \(\pi\) to the target permutation \(\gamma\). Each level of BFS corresponds to one reversal.
- Track the reversals used to transform \(\pi\) to \(\gamma\).
- Implementing BFS:
- Use a queue to manage the permutations to explore.
- Use a set to keep track of visited permutations to avoid redundant work.
- For each permutation, generate all possible permutations resulting from a single reversal and enqueue them if they haven’t been visited.
- Reconstruct Path:
- Once the target permutation \(\gamma\) is reached, backtrack to reconstruct the sequence of reversals.
52.6 Example Solution in Python
Here’s the implementation of the approach:
from collections import deque
def reverse_sublist(lst, start, end):
lst[start:end+1] = lst[start:end+1][::-1]
def bfs(start, target):
queue = deque([(start, [])])
visited = set()
visited.add(tuple(start))
while queue:
current, path = queue.popleft()
if current == target:
return len(path), path
for i in range(len(current)):
for j in range(i+1, len(current)):
new_perm = current[:]
reverse_sublist(new_perm, i, j)
new_tuple = tuple(new_perm)
if new_tuple not in visited:
visited.add(new_tuple)
queue.append((new_perm, path + [(i+1, j+1)]))
return -1, []
def sorting_by_reversals(pi, gamma):
distance, reversals = bfs(pi, gamma)
return distance, reversals
def parse_input(input_string):
lines = input_string.strip().split("\n")
pi = [int(x) for x in lines[0].split()]
gamma = [int(x) for x in lines[1].split()]
return pi, gamma
# Sample input
sample_input = """
6 5 4 7 2 3 9 8 10 1
4 6 2 9 7 1 3 8 5 10
"""
pi, gamma = parse_input(sample_input)
# Get the results
distance, reversals = sorting_by_reversals(pi, gamma)
# Print the results
print(distance)
for r in reversals:
print(r[0], r[1])52.7 Explanation
- reverse_sublist: A helper function to reverse a sublist within a list.
- bfs: The BFS function to explore all possible permutations resulting from single reversals, tracking the path taken.
- sorting_by_reversals: Main function to find the reversal distance and the specific reversals.
53 Inferring Protein from Spectrum
The prefix spectrum of a weighted string is the collection of all its prefix weights.
Given: A list \(L\) of \(n\) (\(n≤100\)) positive real numbers.
Return: A protein string of length \(n−1\) whose prefix spectrum is equal to \(L\) (if multiple solutions exist, you may output any one of them). Consult the monoisotopic mas table.
53.1 Sample Dataset
3524.8542
3710.9335
3841.974
3970.0326
4057.064653.2 Sample Output
WMQS53.3 Solution
To solve this problem, we need to infer a protein string from its prefix spectrum. The prefix spectrum is a list of cumulative masses of prefixes of the protein. Given a list of masses, our goal is to determine which amino acids correspond to the differences between successive masses in this list.
Here’s a step-by-step approach to solve the problem:
- Parse the input list of masses.
- Compute the differences between successive masses. These differences should correspond to the masses of amino acids.
- Match the computed differences to the known monoisotopic masses of amino acids.
- Construct the protein string from the matched amino acids.
We’ll use the monoisotopic mas table for amino acids, which provides the exact masses of each amino acid.
Here’s the Python code to implement this:
# Monoisotopic mas table for amino acids
monoisotopic_mass_table = {
'A': 71.03711, 'C': 103.00919, 'D': 115.02694, 'E': 129.04259, 'F': 147.06841,
'G': 57.02146, 'H': 137.05891, 'I': 113.08406, 'K': 128.09496, 'L': 113.08406,
'M': 131.04049, 'N': 114.04293, 'P': 97.05276, 'Q': 128.05858, 'R': 156.10111,
'S': 87.03203, 'T': 101.04768, 'V': 99.06841, 'W': 186.07931, 'Y': 163.06333
}
def parse_input(input_string):
return list(map(float, input_string.strip().split()))
def find_amino_acid(delta_mass):
for amino_acid, mas in monoisotopic_mass_table.items():
if abs(mas - delta_mass) < 0.01: # Allowing a small tolerance
return amino_acid
return None
def infer_protein_from_spectrum(spectrum):
protein = ""
for i in range(1, len(spectrum)):
delta_mas = spectrum[i] - spectrum[i-1]
amino_acid = find_amino_acid(delta_mass)
if amino_acid:
protein += amino_acid
else:
raise ValueError(f"No matching amino acid found for mas difference {delta_mass}")
return protein
def main():
# Sample input
sample_input = """
3524.8542
3710.9335
3841.974
3970.0326
4057.0646
"""
spectrum = parse_input(sample_input)
protein = infer_protein_from_spectrum(spectrum)
print(protein)
if __name__ == "__main__":
main()53.4 Explanation
- parse_input: This function parses the input string into a list of floats representing the masses.
- find_amino_acid: This function takes a mas difference and finds the corresponding amino acid by comparing it against the monoisotopic mas table, allowing for a small tolerance due to floating-point precision issues.
- infer_protein_from_spectrum: This function computes the differences between successive masses in the spectrum and uses
find_amino_acidto map these differences to amino acids, constructing the protein string.
54 Introduction to Pattern Matching
Given a collection of strings, their trie (often pronounced “try” to avoid ambiguity with the general term tree) is a rooted tree formed as follows. For every unique first symbol in the strings, an edge is formed connecting the root to a new vertex. This symbol is then used to label the edge.
We may then iterate the proces by moving down one level as follows. Say that an edge connecting the root to a node \(v\) is labeled with ‘A’; then we delete the first symbol from every string in the collection beginning with ‘A’ and then treat \(v\) as our root. We apply this proces to all nodes that are adjacent to the root, and then we move down another level and continue.
As a result of this method of construction, the symbols along the edges of any path in the trie from the root to a leaf will spell out a unique string from the collection, as long as no string is a prefix of another in the collection (this would cause the first string to be encoded as a path terminating at an internal node).
Given: A list of at most 100 DNA strings of length at most 100 bp, none of which is a prefix of another.
Return: The adjacency list corresponding to the trie \(T\) for these patterns, in the following format. If \(T\) has \(n\) nodes, first label the root with 1 and then label the remaining nodes with the integers 2 through \(n\) in any order you like. Each edge of the adjacency list of \(T\) will be encoded by a triple containing the integer representing the edge’s parent node, followed by the integer representing the edge’s child node, and finally the symbol labeling the edge.
54.1 Sample Dataset
ATAGA
ATC
GAT54.2 Sample Output
1 2 A
2 3 T
3 4 A
4 5 G
5 6 A
3 7 C
1 8 G
8 9 A
9 10 T54.3 Solution
To construct a trie from a given collection of DNA strings, we need to follow these steps:
- Initialization: Start with a root node.
- Insertion: For each string in the collection, insert it into the trie by creating new nodes and edges as necessary.
- Output: Generate the adjacency list representation of the trie.
Here is the Python code to accomplish this task:
def build_trie(patterns):
trie = {1: {}}
next_node = 2
for pattern in patterns:
current_node = 1
for char in pattern:
if char in trie[current_node]:
current_node = trie[current_node][char]
else:
trie[current_node][char] = next_node
trie[next_node] = {}
current_node = next_node
next_node += 1
return trie
def trie_to_adjacency_list(trie):
adjacency_list = []
for parent in trie:
for char, child in trie[parent].items():
adjacency_list.append((parent, child, char))
return adjacency_list
def main():
# Sample input
input_data = """
ATAGA
ATC
GAT
"""
patterns = input_data.strip().split()
# Build the trie
trie = build_trie(patterns)
# Convert trie to adjacency list
adjacency_list = trie_to_adjacency_list(trie)
# Print the adjacency list
for parent, child, char in adjacency_list:
print(f"{parent} {child} {char}")
if __name__ == "__main__":
main()54.4 Explanation
- Function
build_trie:- Initializes the trie with a root node labeled
1. - For each pattern, it iterates through its characters, and either moves to an existing node if the character is already in the current node’s dictionary or creates a new node if the character is not present.
- It maintains a counter
next_nodeto assign new labels to nodes.
- Initializes the trie with a root node labeled
- Function
trie_to_adjacency_list:- Converts the trie structure into an adjacency list format, which is a list of tuples where each tuple represents an edge from a parent node to a child node labeled by a character.
- Function
main:- Handles input and output operations.
- Reads the input strings, constructs the trie, converts it to an adjacency list, and then prints the adjacency list.
55 Comparing Spectra with the Spectral Convolution
A multiset is a generalization of the notion of set to include a collection of objects in which each object may occur more than once (the order in which objects are given is still unimportant). For a multiset \(S\), the multiplicity of an element \(x\) is the number of times that \(x\) occurs in the set; this multiplicity is denoted \(S(x)\). Note that every set is included in the definition of multiset.
The Minkowski sum of multisets \(S1\) and \(S2\) containing real numbers is the new multiset \(S1⊕S2\) formed by taking all possible sums \(s1+s2\) of an element \(s1\) from \(S1\) and an element \(s2\) from \(S2\). The Minkowski sum could be defined more concisely as \(S1⊕S2=s1+s2:s1∈S1,s2∈S2\), The Minkowski difference \(S1⊖S2\) is defined analogously by taking all possible differences \(s1−s2\).
If \(S1\) and \(S2\) represent simplified spectra taken from two peptides, then \(S1⊖S2\) is called the spectral convolution of \(S1\) and \(S2\). In this notation, the shared peaks count is represented by \((S2⊖S1)(0)\), and the value of \(x\) for which \((S2⊖S1)(x)\) has the maximal value is the shift value maximizing the number of shared masses of \(S1\) and \(S2\).
Given: Two multisets of positive real numbers \(S1\) and \(S2\). The size of each multiset is at most 200.
Return: The largest multiplicity of \(S1⊖S2\), as well as the absolute value of the number \(x\) maximizing \((S1⊖S2)(x)\) (you may return any such value if multiple solutions exist).
55.1 Sample Dataset
186.07931 287.12699 548.20532 580.18077 681.22845 706.27446 782.27613 968.35544 968.35544
101.04768 158.06914 202.09536 318.09979 419.14747 463.1736955.2 Sample Output
3
85.0316355.3 Solution
To solve the problem of comparing spectra using the spectral convolution, we need to follow these steps:
- Parse the input: Read the two multisets \(S1\) and \(S2\).
- Compute the spectral convolution: For each pair of elements \((s1, s2)\) where \(s1 \in S1\) and \(s2 \in S2\), compute the difference \(s1 - s2\).
- Count the occurrences of each difference: Track how many times each difference appears.
- Find the most frequent difference: Identify the difference that appears most frequently and its multiplicity.
Here’s the Python code to implement the above steps:
from collections import Counter
def parse_input(input_data):
lines = input_data.strip().split("\n")
S1 = list(map(float, lines[0].split()))
S2 = list(map(float, lines[1].split()))
return S1, S2
def spectral_convolution(S1, S2):
convolution = []
for s1 in S1:
for s2 in S2:
convolution.append(round(s1 - s2, 5))
return convolution
def find_max_multiplicity(convolution):
count = Counter(convolution)
max_value, max_count = max(count.items(), key=lambda x: x[1])
return max_count, abs(max_value)
def main(input_data):
S1, S2 = parse_input(input_data)
convolution = spectral_convolution(S1, S2)
max_count, max_value = find_max_multiplicity(convolution)
return max_count, max_value
# Sample input
sample_input = """
186.07931 287.12699 548.20532 580.18077 681.22845 706.27446 782.27613 968.35544 968.35544
101.04768 158.06914 202.09536 318.09979 419.14747 463.17369
"""
# Proces the input and get the result
result = main(sample_input)
print(result[0])
print(f"{result[1]:f}")55.4 Explanation
- Parsing Input:
- The
parse_inputfunction reads the input data, splits it into lines, and then converts each line into a list of floats representing \(S1\) and \(S2\).
- The
- Computing Spectral Convolution:
- The
spectral_convolutionfunction takes all pairs \((s1, s2)\) from \(S1\) and \(S2\), computes the difference \(s1 - s2\), and stores these differences in a list. Theroundfunction ensures precision to 5 decimal places, as floating-point arithmetic can introduce small errors.
- The
- Counting Occurrences:
- The
find_max_multiplicityfunction uses Python’sCounterfrom thecollectionsmodule to count how often each difference appears in the convolution list. It then finds the difference with the maximum count (multiplicity) and its corresponding value.
- The
- Main Function:
- The
mainfunction orchestrates the proces by calling the helper functions and printing the result.
- The
56 Creating a Character Table
Given a collection of \(n\) taxa, any subset \(S\) of these taxa can be seen as encoding a character that divides the taxa into the sets \(S\) and \(S^c\); we can represent the character by \(S∣S^c\), which is called a split. Alternately, the character can be represented by a character array \(A\) of length \(n\) for which \(A[j]=1\) if the \(j\)th taxon belongs to \(S\) and \(A[j]=0\) if the \(j\)th taxon belongs to \(S^c\) (recall the “ON”/“OFF” analogy from “Counting Subsets”).
At the same time, observe that the removal of an edge from an unrooted binary tree produces two separate trees, each one containing a subset of the original taxa. So each edge may also be encoded by a split \(S∣S^c\).
A trivial character isolates a single taxon into a group of its own. The corresponding split \(S∣S^c\) must be such that \(S\) or \(S^c\) contains only one element; the edge encoded by this split must be incident to a leaf of the unrooted binary tree, and the array for the character contains exactly one 0 or exactly one 1. Trivial characters are of no phylogenetic interest because they fail to provide us with information regarding the relationships of taxa to each other. All other characters are called nontrivial characters (and the associated splits are called nontrivial splits).
A character table is a matrix \(C\) in which each row represents the array notation for a nontrivial character. That is, entry \(C_{i,j}\) denotes the “ON”/“OFF” position of the \(i\)th character with respect to the \(j\)th taxon.
Given: An unrooted binary tree \(T\) in Newick format for at most 200 species taxa.
Return: A character table having the same splits as the edge splits of \(T\). The columns of the character table should encode the taxa ordered lexicographically; the rows of the character table may be given in any order. Also, for any given character, the particular subset of taxa to which 1s are assigned is arbitrary.
56.1 Sample Dataset
(dog,((elephant,mouse),robot),cat);56.2 Sample Output
00110
0011156.3 Solution
The code parses a Newick string representing a phylogenetic tree and converts it into a character table, where each row of the table represents a partition of taxa.
from collections import defaultdict
# Node clas to represent a node in the tree
clas Node:
def __init__(self, name=""):
self.name = name # Name of the taxon or internal node
self.children = [] # List to store child nodes
# Function to parse a Newick string into a tree structure
def parse_newick(newick):
def parse_node():
nonlocal i
node = Node()
if newick[i] == '(':
i += 1
while newick[i] != ')':
node.children.append(parse_node())
if newick[i] == ',':
i += 1
i += 1
# Extract node name
if newick[i] not in ',)':
name_start = i
while newick[i] not in ',)':
i += 1
node.name = newick[name_start:i]
return node
i = 0
return parse_node()
# Recursive function to find all splits (partitions) of the taxa
def find_splits(node, taxa, splits):
# If it's a leaf node, return the set containing the taxon name
if not node.children:
return {node.name}
# Recursively find splits in left and right children
left = find_splits(node.children[0], taxa, splits)
right = find_splits(node.children[1], taxa, splits)
# Combine left and right splits
split = left | right
# Check if this split is non-trivial and add it to the splits list
if 1 < len(split) < len(taxa) - 1:
splits.append(split)
return split
# Function to create a character table from the splits
def create_character_table(tree, taxa):
splits = []
find_splits(tree, set(taxa), splits)
table = []
# Convert each split into a binary row
for split in splits:
row = ['1' if taxon in split else '0' for taxon in taxa]
table.append(''.join(row))
return table
# Sample Newick string
sample_input = """
(dog,((elephant,mouse),robot),cat);
"""
# Strip leading/trailing whitespace and parse the Newick string
newick = sample_input.strip()
tree = parse_newick(newick)
taxa = []
# Function to collect all taxa names from the tree
def collect_taxa(node):
if node.name:
taxa.append(node.name)
for child in node.children:
collect_taxa(child)
# Collect and sort taxa names
collect_taxa(tree)
taxa.sort()
# Create the character table based on the collected taxa
character_table = create_character_table(tree, taxa)
# Print each row of the character table
for row in character_table:
print(row)56.4 Explain
NodeClass:- Purpose: Represents a node in the tree.
- Attributes:
name: Name of the taxon or internal node.children: List of child nodes.
parse_newickFunction:- Purpose: Parses a Newick string into a tree structure.
- Inner Function
parse_node:- Handles parentheses: It processes nested parentheses to build the tree structure.
- Extracts names: It extracts the name of each node by looking for characters until it hits a delimiter (comma or closing parenthesis).
find_splitsFunction:- Purpose: Finds and collects all non-trivial splits of the taxa.
- Parameters:
node: Current node in the tree.taxa: Set of all taxa names.splits: List to collect all non-trivial splits.
- Logic:
- Recursively computes splits for left and right subtrees.
- Combines splits and checks if they are non-trivial.
- Adds valid splits to the
splitslist.
create_character_tableFunction:- Purpose: Converts splits into a character table.
- Parameters:
tree: Root node of the tree.taxa: List of sorted taxa names.
- Logic:
- Uses
find_splitsto get the splits. - Converts each split into a binary representation.
- Constructs and returns the character table as a list of strings.
- Uses
- Main Execution:
- Input Handling: Reads and strips the Newick string, then parses it into a tree.
- Taxa Collection: Collects and sorts all taxa names from the tree.
- Character Table Creation: Generates and prints the character table based on the tree structure and taxa.
57 Constructing a De Bruijn Graph
Consider a set \(S\) of \((k+1)\)-mers of some unknown DNA string. Let Src𝑆rc denote the set containing all reverse complements of the elements of \(S\). (recall from “Counting Subsets” that sets are not allowed to contain duplicate elements).
The de Bruijn graph Bk𝐵𝑘 of order \(k\) corresponding to \(S∪S^{rc}\) is a digraph defined in the following way:
- Nodes of \(B_k\) correspond to all \(k\)-mers that are present as a substring of a \((k+1)\)-mer from \(S∪S^{rc}\).
- Edges of \(B_k\) are encoded by the \((k+1)\)-mers of \(S∪S^{rc}\) in the following way: for each \((k+1)\)-mer \(r\) in \(S∪S^{rc}\), form a directed edge (\(r[1:k]\), \(r[2:k+1]\)).
Given: A collection of up to 1000 (possibly repeating) DNA strings of equal length (not exceeding 50 bp) corresponding to a set \(S\) of \((k+1)\)-mers.
Return: The adjacency list corresponding to the de Bruijn graph corresponding to \(S∪S^{rc}\).
57.1 Sample Dataset
TGAT
CATG
TCAT
ATGC
CATC
CATC57.2 Sample Output
(ATC, TCA)
(ATG, TGA)
(ATG, TGC)
(CAT, ATC)
(CAT, ATG)
(GAT, ATG)
(GCA, CAT)
(TCA, CAT)
(TGA, GAT)57.3 Solution
We’ll compute the reverse complements and the updated set separately and then proceed with constructing the De Bruijn graph.
from collections import defaultdict
def reverse_complement(dna):
"""Computes the reverse complement of a DNA string."""
complement = {'A': 'T', 'T': 'A', 'C': 'G', 'G': 'C'}
return ''.join(complement[base] for base in reversed(dna))
def construct_de_bruijn_graph(kmers):
"""Constructs the De Bruijn graph and returns the adjacency list."""
# Create a new set to include reverse complements
kmers_with_rc = set(kmers)
for kmer in kmers:
rc_kmer = reverse_complement(kmer)
kmers_with_rc.add(rc_kmer)
adjacency_list = defaultdict(set)
k = len(next(iter(kmers))) - 1 # Length of the k-mer
for kmer in kmers_with_rc:
for i in range(len(kmer) - k):
prefix = kmer[i:i+k]
suffix = kmer[i+1:i+k+1]
adjacency_list[prefix].add(suffix)
return adjacency_list
def format_adjacency_list(adj_list):
"""Formats the adjacency list into the required output format."""
result = []
for start_node, end_nodes in adj_list.items():
for end_node in end_nodes:
result.append(f"({start_node}, {end_node})")
return sorted(result)
def main(input_data):
"""Main function to proces the input data and generate the De Bruijn graph."""
lines = input_data.strip().split('\n')
kplus1_mers = set(lines)
# Determine k from the length of the (k+1)-mers
k = len(next(iter(kplus1_mers))) - 1
# Construct the De Bruijn graph
adj_list = construct_de_bruijn_graph(kplus1_mers)
# Format and print the adjacency list
formatted_output = format_adjacency_list(adj_list)
for line in formatted_output:
print(line)
# Sample input
sample_input = """
TGAT
CATG
TCAT
ATGC
CATC
CATC
"""
# Run the main function with the sample input
main(sample_input)57.4 Changes Made
- Avoided Modifying Set During Iteration:
- Instead of modifying
kmerswhile iterating over it, we create a new setkmers_with_rcthat initially contains all the originalkmersand then add reverse complements to it.
- Instead of modifying
- Fixed the Extraction of Prefix and Suffix:
- Adjusted the slicing in the De Bruijn graph construction to ensure we correctly extract the prefix and suffix (k)-mers from each (k+1)-mer.
- Ensured Proper Handling of Adjacency List:
- Correctly formatted the adjacency list to meet the output requirements.
58 Edit Distance Alignment
An alignment of two strings s and t is defined by two strings s′ and t′ satisfying the following three conditions: 1. s′ and t′ must be formed from adding gap symbols “-” to each of s and t, respectively; as a result, s and t will form subsequences of s′ and t′. 2. s′ and t′ must have the same length. 3. Two gap symbols may not be aligned; that is, if s′[j] is a gap symbol, then t′[j] cannot be a gap symbol, and vice-versa.
We say that s′ and t′ augment s and t. Writing s′ directly over t′ so that symbols are aligned provides us with a scenario for transforming s into t. Mismatched symbols from s and t correspond to symbol substitutions; a gap symbol s′[j] aligned with a non-gap symbol t′[j] implies the insertion of this symbol into t; a gap symbol t′[j] aligned with a non-gap symbol s′[j] implies the deletion of this symbol from s.
Thus, an alignment represents a transformation of s into t via edit operations. We define the corresponding edit alignment score of s′ and t′ as \(dH(s′,t′)\) (Hamming distance is used because the gap symbol has been introduced for insertions and deletions). It follows that \(dE(s,t)=mins′,t′dH(s′,t′)\), where the minimum is taken over all alignments of s and t𝑡. We call such a minimum score alignment an optimal alignment (with respect to edit distance).
Given: Two protein strings s and t in FASTA format (with each string having length at most 1000 aa).
Return: The edit distance \(dE(s,t)\) followed by two augmented strings s′ and t′ representing an optimal alignment of s and t.
58.1 Sample Dataset
>Rosalind_43
PRETTY
>Rosalind_97
PRTTEIN58.2 Sample Output
4
PRETTY--
PR-TTEIN58.3 Solution
def parse_fasta(fasta_str):
"""
Parse a FASTA format string into a list of sequences.
"""
sequences = []
current_sequence = []
for line in fasta_str.strip().split("\n"):
if line.startswith(">"):
if current_sequence:
sequences.append("".join(current_sequence))
current_sequence = []
else:
current_sequence.append(line.strip())
if current_sequence:
sequences.append("".join(current_sequence))
return sequences
def edit_distance_alignment(s, t):
"""
Compute the edit distance and optimal alignment of two strings.
"""
m, n = len(s), len(t)
dp = [[0] * (n + 1) for _ in range(m + 1)]
# Initialize the dp table for base cases
for i in range(m + 1):
dp[i][0] = i
for j in range(n + 1):
dp[0][j] = j
# Fill the dp table
for i in range(1, m + 1):
for j in range(1, n + 1):
if s[i-1] == t[j-1]:
dp[i][j] = dp[i-1][j-1]
else:
dp[i][j] = min(dp[i-1][j], dp[i][j-1], dp[i-1][j-1]) + 1
# Traceback to construct the aligned strings
s_aligned, t_aligned = "", ""
i, j = m, n
while i > 0 and j > 0:
if s[i-1] == t[j-1]:
s_aligned = s[i-1] + s_aligned
t_aligned = t[j-1] + t_aligned
i -= 1
j -= 1
elif dp[i][j] == dp[i-1][j] + 1:
s_aligned = s[i-1] + s_aligned
t_aligned = "-" + t_aligned
i -= 1
elif dp[i][j] == dp[i][j-1] + 1:
s_aligned = "-" + s_aligned
t_aligned = t[j-1] + t_aligned
j -= 1
else:
s_aligned = s[i-1] + s_aligned
t_aligned = t[j-1] + t_aligned
i -= 1
j -= 1
# Handle any remaining characters
while i > 0:
s_aligned = s[i-1] + s_aligned
t_aligned = "-" + t_aligned
i -= 1
while j > 0:
s_aligned = "-" + s_aligned
t_aligned = t[j-1] + t_aligned
j -= 1
return dp[m][n], s_aligned, t_aligned
# Sample input
sample_input = """
>Rosalind_43
PRETTY
>Rosalind_97
PRTTEIN
"""
# Proces the input
sequences = parse_fasta(sample_input)
s, t = sequences[0], sequences[1]
# Compute edit distance and alignment
edit_distance, s_aligned, t_aligned = edit_distance_alignment(s, t)
# Print the results
print(edit_distance)
print(s_aligned)
print(t_aligned)58.4 Explanation of the Code
parse_fasta(fasta_str): Convert a FASTA format string into a list of sequences.- Split the input string into lines.
- Collect sequence lines into
current_sequenceuntil a new header line is encountered. - Append the complete sequence to
sequenceswhen a new header is found. - Return the list of sequences.
edit_distance_alignment(s, t): Compute the edit distance and provide an optimal alignment of two sequences.- Initialize DP Table: Set up a 2D table
dpwheredp[i][j]holds the minimum edit distance between the firsticharacters ofsand the firstjcharacters oft. - Fill DP Table: Use dynamic programming to calculate the edit distance considering substitutions, insertions, and deletions.
- Traceback: Build the aligned sequences by following the
dptable from the bottom-right to the top-left, handling matches, insertions, and deletions. - Handle Remaining Characters: If there are remaining characters in either string after the traceback, append them with gaps.
- Initialize DP Table: Set up a 2D table
59 Inferring Peptide from Full Spectrum
Say that we have a string \(s\) containing \(t\) as an internal substring, so that there exist nonempty substrings \(s1\) and \(s2\) of \(s\) such that \(s\) can be written as \(s1ts2\). A t-prefix contains all of \(s1\) and none of \(s2\); likewise, a t-suffix contains all of \(s2\) and none of \(s1\).
Given: A list \(L\) containing \(2n+3\) positive real numbers (\(n≤100\)). The first number in \(L\) is the parent mas of a peptide \(P\), and all other numbers represent the masses of some b-ions and y-ions of \(P\) (in no particular order). You may assume that if the mas of a b-ion is present, then so is that of its complementary y-ion, and vice-versa.
Return: A protein string \(t\) of length \(n\) for which there exist two positive real numbers \(w1\) and \(w2\) such that for every prefix p𝑝 and suffix \(s\) of \(t\), each of \(w(p)+w1\) and \(w(s)+w2\) is equal to an element of \(L\). (In other words, there exists a protein string whose \(t\)-prefix and \(t\)-suffix weights correspond to the non-parent mas values of \(L\).) If multiple solutions exist, you may output any one.
59.1 Sample Dataset
1988.21104821
610.391039105
738.485999105
766.492149105
863.544909105
867.528589105
992.587499105
995.623549105
1120.6824591
1124.6661391
1221.7188991
1249.7250491
1377.820009159.2 Sample Output
KEKEP59.3 Solution
import random
from typing import List, Tuple, Dict, Union
# Amino acid mas mapping
amino_acid_masses: Dict[float, List[str]] = {
57.02146: ["G"], 71.03711: ["A"], 87.03203: ["S"], 97.05276: ["P"], 99.06841: ["V"],
101.04768: ["T"], 103.00919: ["C"], 113.08406: ["I", "L"], 114.04293: ["N"], 115.02694: ["D"],
128.05858: ["Q"], 128.09496: ["K"], 129.04259: ["E"], 131.04049: ["M"], 137.05891: ["H"],
147.06841: ["F"], 156.10111: ["R"], 163.06333: ["Y"], 186.07931: ["W"],
}
def infer_peptide(n: int, parent_mass: float, ion_masses: List[float], peptides: List[str]) -> List[str]:
"""
Infers peptide sequences based on given ion masses and the target peptide length.
:param n: Length of the peptide to be inferred.
:param parent_mass: Mas of the parent peptide.
:param ion_masses: List of ion masses representing b-ions and y-ions.
:param peptides: List of current peptide candidates.
:return: List of inferred peptide sequences.
"""
if len(peptides[0]) == n:
return peptides
possible_ions = [] # List to store possible amino acids between ion pairs
# Find possible amino acids between ion pairs
for i in range(len(ion_masses) - 1):
for j in range(i + 1, len(ion_masses)):
delta_mas = round(ion_masses[j] - ion_masses[i], 5)
amino_acids = amino_acid_masses.get(delta_mass, [])
if amino_acids:
possible_ions.append((i, j, amino_acids))
if possible_ions:
# Update ion masses and peptide candidates
new_ion_masses = ion_masses[possible_ions[0][1]:]
new_amino_acids = possible_ions[0][2]
new_peptides = [peptide + aa for peptide in peptides for aa in new_amino_acids]
# Recursively infer peptide sequences
return infer_peptide(n, parent_mass, new_ion_masses, new_peptides)
return peptides
# Sample input
sample_input = """
1988.21104821
610.391039105
738.485999105
766.492149105
863.544909105
867.528589105
992.587499105
995.623549105
1120.6824591
1124.6661391
1221.7188991
1249.7250491
1377.8200091
"""
# Parse input data
input_lines = [float(line) for line in sample_input.strip().split("\n")]
parent_mass, ion_masses = input_lines[0], input_lines[1:]
# Determine the length of the peptide
peptide_length = (len(ion_masses) - 2) // 2
# Infer peptide sequences
possible_peptides = infer_peptide(peptide_length, parent_mass, ion_masses, [""])
# Print a random peptide sequence
print(random.choice(possible_peptides))60 Independent Segregation of Chromosomes
Consider a collection of coin flips. One of the most natural questions we can ask is if we flip a coin 92 times, what is the probability of obtaining 51 “heads”, vs. 27 “heads”, vs. 92 “heads”?
Each coin flip can be modeled by a uniform random variable in which each of the two outcomes (“heads” and “tails”) has probability equal to \(1/2\). We may assume that these random variables are independent (see “Independent Alleles”); in layman’s terms, the outcomes of the two coin flips do not influence each other.
A binomial random variable \(X\) takes a value of \(k\) if \(n\) consecutive “coin flips” result in \(k\) total “heads” and \(n−k\) total “tails.” We write that \(X∈Bin(n/2)\).
Given: A positive integer \(n≤50\).
Return: An array \(A\) of length \(2n\) in which \(A[k]\) represents the common logarithm of the probability that two diploid siblings share at least \(k\) of their \(2n\) chromosomes (we do not consider recombination for now).
60.1 Sample Dataset
560.2 Sample Output
0.000 -0.005 -0.024 -0.082 -0.206 -0.424 -0.765 -1.262 -1.969 -3.01060.3 Solution
import math
def calculate_shared_chromosome_probabilities(sample_input: str):
"""
Calculate the common logarithm of the probability that two diploid siblings
share at least k of their 2n chromosomes, given n.
Args:
- sample_input (str): The input string representing the value of n.
Returns:
- List of float: Logarithm base 10 of the cumulative probabilities.
"""
# Parse the sample input to an integer
n = int(sample_input.strip())
# Probability of sharing each chromosome (independent coin flip)
p = 0.5
# Initialize the cumulative probability and the result array
Pr = 0
A = []
# Loop from 2*n down to 1 (inclusive) to calculate cumulative probabilities
for k in range(2 * n, 0, -1):
# Calculate the binomial coefficient: C(2n, k)
binom_coeff = math.factorial(2 * n) / (math.factorial(k) * math.factorial(2 * n - k))
# Calculate the probability of exactly k shared chromosomes
Pr += binom_coeff * math.pow(p, k) * math.pow(1 - p, 2 * n - k)
# Append the common logarithm (base 10) of the cumulative probability to the result array
A.append(math.log10(Pr))
# Return the result array in reverse order
return [round(value, 3) for value in A[::-1]]
# Example usage
sample_input = "5"
result = calculate_shared_chromosome_probabilities(sample_input)
print(" ".join(f"{value:3f}" for value in result))60.4 Explanation of the Function
- Function Definition:
- The function
calculate_shared_chromosome_probabilitiestakes a stringsample_input.
- The function
- Parse Input:
sample_inputis stripped of any surrounding whitespace and converted to an integern.
- Initialize Variables:
pis set to 0.5, representing the probability of sharing each chromosome.Pris initialized to store the cumulative probability.Ais an empty list to store the logarithms of cumulative probabilities.
- Calculate Cumulative Probabilities:
- Loop from
2*ndown to1to calculate the cumulative probability for at leastkshared chromosomes. - For each
k, compute the binomial coefficient \(C(2n, k)\). - Calculate the probability of exactly
kshared chromosomes and add it toPr. - Append the logarithm (base 10) of
Prto the listA.
- Loop from
- Return the Result:
- Return the values in
Ain reverse order, rounded to 3 decimal places.
- Return the values in
- Example Usage:
- The function is called with a sample input
"5", and the results are printed in the specified format.
- The function is called with a sample input
61 Finding Disjoint Motifs in a Gene
Given three strings \(s\), \(t\), and \(u\), we say that \(t\) and \(u\) can be interwoven into \(s\) if there is some substring of \(s\) made up of \(t\) and \(u\) as disjoint subsequences.
For example, the strings “ACAGACAG” and “CCGCCG” can be interwoven into “GACCACGGTTGACCACGGTT”. However, they cannot be interwoven into “GACCACAAAAGGTTGACCACAAAAGGTT” because of the appearance of the four ’A’s in the middle of the subsequences. Similarly, even though both “ACACGACACG” is a shortest common supersequence of ACAGACAG and CCGCCG, it is not possible to interweave these two strings into “ACACGACACG” because the two desired subsequences must be disjoint; see “Interleaving Two Motifs” for details on finding a shortest common supersequence of two strings.
Given: A text DNA string \(s\) of length at most 10 kbp, followed by a collection of \(n\) (\(n≤10\)) DNA strings of length at most 10 bp acting as patterns.
Return: An \(n×n\) matrix \(M\) for which \(M_{j,k}=1\) if the \(j\)th and \(k\)th pattern strings can be interwoven into \(s\) and \(M_{j,k}=0\) otherwise.
61.1 Sample Dataset
GACCACGGTT
ACAG
GT
CCG61.2 Sample Output
0 0 1
0 1 0
1 0 061.3 Solution
import numpy as np
def is_interwoven(dna1, dna2, superstr):
"""
Recursive function to check if dna1 and dna2 can be interwoven to form superstr.
"""
if len(superstr) == 0:
return True
elif dna1 and dna2 and dna1[0] == dna2[0] == superstr[0]:
return is_interwoven(dna1[1:], dna2, superstr[1:]) or is_interwoven(dna1, dna2[1:], superstr[1:])
elif dna1 and dna1[0] == superstr[0]:
return is_interwoven(dna1[1:], dna2, superstr[1:])
elif dna2 and dna2[0] == superstr[0]:
return is_interwoven(dna1, dna2[1:], superstr[1:])
else:
return False
def find_disjoint_motifs(super_string, patterns):
"""
Function to find the disjoint motifs matrix for the given super_string and patterns.
"""
n = len(patterns)
M = np.zeros((n, n), dtype=int)
for i in range(n):
for j in range(i, n):
pattern1 = patterns[i]
pattern2 = patterns[j]
combined_length = len(pattern1) + len(pattern2)
combined_profile = [pattern1.count(nuc) + pattern2.count(nuc) for nuc in "ACGT"]
for index in range(len(super_string) - combined_length + 1):
superstr_segment = super_string[index:index + combined_length]
superstr_profile = [superstr_segment.count(nuc) for nuc in "ACGT"]
if combined_profile == superstr_profile:
if is_interwoven(pattern1 + '$', pattern2 + '$', superstr_segment):
M[i][j] = 1
break
if i != j:
M[j][i] = M[i][j]
return M
# Sample dataset
sample_input = """
GACCACGGTT
ACAG
GT
CCG
"""
data = sample_input.strip().split()
super_string = data[0]
patterns = data[1:]
# Find the disjoint motifs matrix
result_matrix = find_disjoint_motifs(super_string, patterns)
# Print the result matrix
for row in result_matrix:
print(" ".join(map(str, row)))61.4 Explanation
- is_interwoven Function:
- Purpose: To check if
dna1anddna2can be interwoven to formsuperstr. - Parameters:
dna1,dna2, andsuperstr. - Logic:
- If
superstris empty, returnTruebecause the interweaving is complete. - If both
dna1anddna2are non-empty and their first characters match the first character ofsuperstr, recursively check both possibilities (taking fromdna1ordna2). - If the first character of
dna1matches the first character ofsuperstr, recursively check the remaining parts. - If the first character of
dna2matches the first character ofsuperstr, recursively check the remaining parts. - If none of the above conditions are met, return
False.
- If
- Purpose: To check if
- find_disjoint_motifs Function:
- Purpose: To find the disjoint motifs matrix for the given
super_stringandpatterns. - Parameters:
super_stringandpatterns. - Logic:
- Initialize a zero matrix
Mof sizen x nwherenis the number of patterns. - For each pair of patterns
pattern1andpattern2, calculate their combined length and nucleotide profile. - Iterate over all possible substrings of
super_stringof the same length. - Compare the nucleotide profile of the substring with the combined profile.
- If they match, use
is_interwovento check if they can be interwoven to form the substring. - Update the matrix
Maccordingly. - Since the comparison is symmetric, update both
M[i][j]andM[j][i].
- Initialize a zero matrix
- Purpose: To find the disjoint motifs matrix for the given
- Main Execution:
- Parse the input to extract the
super_stringandpatterns. - Call
find_disjoint_motifsto get the result matrix. - Print the result matrix.
- Parse the input to extract the
62 Finding the Longest Multiple Repeat
A repeated substring of a string \(s\) of length \(n\) is simply a substring that appears in more than one location of \(s\); more specifically, a k-fold substring appears in at least k distinct locations.
The suffix tree of \(s\), denoted \(T(s)\), is defined as follows:
- \(T(s)\) is a rooted tree having exactly n leaves.
- Every edge of \(T(s)\) is labeled with a substring of \(s^∗\), where \(s^∗\) is the string formed by adding a placeholder symbol
$to the end of \(s\). - Every internal node of \(T(s)\) other than the root has at least two children; i.e., it has degree at least 3.
- The substring labels for the edges leading from a node to its children must begin with different symbols.
- By concatenating the substrings along edges, each path from the root to a leaf corresponds to a unique suffix of \(s^∗\).
Given: A DNA string s (of length at most 20 kbp) with $ appended, a positive integer kk, and a list of edges defining the suffix tree of ss. Each edge is represented by four components:
- the label of its parent node in \(T(s)\);
- the label of its child node in \(T(s)\);
- the location of the substring \(t\) of \(s^∗\) assigned to the edge; and the length of \(t\).
Return: The longest substring of s that occurs at least \(k\) times in \(s\). (If multiple solutions exist, you may return any single solution.)
62.1 Sample Dataset
CATACATAC$
2
node1 node2 1 1
node1 node7 2 1
node1 node14 3 3
node1 node17 10 1
node2 node3 2 4
node2 node6 10 1
node3 node4 6 5
node3 node5 10 1
node7 node8 3 3
node7 node11 5 1
node8 node9 6 5
node8 node10 10 1
node11 node12 6 5
node11 node13 10 1
node14 node15 6 5
node14 node16 10 162.2 Sample Output
CATAC62.3 Solution
To solve this problem, we need to find the longest substring that appears at least k times in a given string s, using its suffix tree. Here’s how we can approach the problem step-by-step:
62.4 Steps to Approach the Problem
- Parse the Input:
- Read the DNA string
swith$appended. - Read the integer
k. - Parse the edges defining the suffix tree.
- Read the DNA string
- Suffix Tree Representation:
- Use the given edges to construct the suffix tree.
- Each edge contains information about the parent node, child node, start position of the substring in
s, and the length of the substring.
- Traverse the Suffix Tree:
- Traverse the tree to count the occurrences of substrings.
- Use a depth-first search (DFS) to explore all paths from the root to the leaves.
- Identify the Longest Substring with at least
kOccurrences:- Track the longest substring that meets the condition during the traversal.
62.5 Implementation
Here’s the Python code to implement the solution:
clas SuffixTreeNode:
def __init__(self):
self.children = {}
self.start = -1
self.length = -1
self.parent = None
def build_suffix_tree(edges, s):
nodes = {}
for edge in edges:
parent, child, start, length = edge
if parent not in nodes:
nodes[parent] = SuffixTreeNode()
if child not in nodes:
nodes[child] = SuffixTreeNode()
nodes[child].parent = nodes[parent]
nodes[child].start = start - 1 # Convert to zero-based index
nodes[child].length = length
nodes[parent].children[child] = nodes[child]
return nodes
def dfs(node, s, k, path, results):
if len(node.children) == 0:
return 1 # Leaf node
count = 0
for child in node.children.values():
count += dfs(child, s, k, path + s[child.start:child.start + child.length], results)
if count >= k:
results.append((path, len(path)))
return count
def longest_k_fold_substring(s, k, edges):
nodes = build_suffix_tree(edges, s)
root = nodes['node1']
results = []
dfs(root, s, k, "", results)
results.sort(key=lambda x: x[1], reverse=True)
return results[0][0] if results else ""
# Sample input
sample_input = """
CATACATAC$
2
node1 node2 1 1
node1 node7 2 1
node1 node14 3 3
node1 node17 10 1
node2 node3 2 4
node2 node6 10 1
node3 node4 6 5
node3 node5 10 1
node7 node8 3 3
node7 node11 5 1
node8 node9 6 5
node8 node10 10 1
node11 node12 6 5
node11 node13 10 1
node14 node15 6 5
node14 node16 10 1
"""
data = sample_input.strip().split('\n')
s = data[0]
k = int(data[1])
edges = [tuple(line.split()) for line in data[2:]]
edges = [(e[0], e[1], int(e[2]), int(e[3])) for e in edges]
result = longest_k_fold_substring(s, k, edges)
print(result)62.6 Explanation
- SuffixTreeNode Class:
- A clas to represent each node in the suffix tree.
- build_suffix_tree Function:
- Constructs the suffix tree using the given edges.
- dfs Function:
- Performs a depth-first search to count the occurrences of substrings and keep track of the valid ones.
- longest_k_fold_substring Function:
- Builds the suffix tree, performs DFS, and identifies the longest substring with at least
koccurrences.
- Builds the suffix tree, performs DFS, and identifies the longest substring with at least
- Sample Input:
- Parses the input and invokes the
longest_k_fold_substringfunction to find and print the result.
- Parses the input and invokes the
63 Newick Format with Edge Weights
In a weighted tree, each edge is assigned a (usually positive) number, called its weight. The distance between two nodes in a weighted tree becomes the sum of the weights along the unique path connecting the nodes.
To generalize Newick format to the case of a weighted tree \(T\), during our repeated “key step,” if leaves \(v_1,v_2,…,v_n\) are neighbors in \(T\), and all these leaves are incident to \(u\), then we replace uu with \((v_1:d_1,v_2:d_2,…,v_n:d_n)u\), where didi is now the weight on the edge \({v_i,u}\).
Given: A collection of \(n\) weighted trees (\(n≤40\)) in Newick format, with each tree containing at most 200 nodes; each tree \(T_k\) is followed by a pair of nodes \(x_k\) and \(y_k\) in \(T_k\).
Return: A collection of n numbers, for which the \(k\)th number represents the distance between \(x_k\) and \(y_k\) in \(T_k\).
63.1 Sample Dataset
(dog:42,cat:33);
cat dog
((dog:4,cat:3):74,robot:98,elephant:58);
dog elephant63.2 Sample Output
75 13663.3 Solution
clas Node:
def __init__(self, number, parent, name=None):
self.number = number
self.parent = parent
self.name = "Node_" + str(self.number) if name is None else name
def __repr__(self):
tmp = ""
if self.name != "Node_" + str(self.number):
tmp = f"({self.name})"
return f"Node_{self.number}{tmp}"
clas WeightedNewick:
def __init__(self, data):
self.nodes = []
self.edge_weight = {}
self.construct_tree(data)
self.name_index = {node.name: node.number for node in self.nodes}
def construct_tree(self, data):
"""Constructs the Newick Tree from the input data."""
data = data.replace(',', ' ').replace('(', '( ').replace(')', ' )').strip(';').split()
current_parent = Node(-1, None)
for item in data:
if item[0] == '(':
# New internal node
current_parent = Node(len(self.nodes), current_parent.number)
self.nodes.append(current_parent)
elif item[0] == ')':
# End of a subtree, backtrack to parent
if len(item) > 1:
self.edge_weight[(current_parent.number, current_parent.parent)] = int(item[item.find(':') + 1:])
if len(item) > 2:
current_parent.name = item[1:item.find(':')]
current_parent = self.nodes[current_parent.parent]
else:
# Leaf node
self.edge_weight[(len(self.nodes), current_parent.number)] = int(item[item.find(':') + 1:])
self.nodes.append(Node(len(self.nodes), current_parent.number, item[:item.find(':')]))
def distance(self, name1, name2):
"""Returns the distance between nodes with names name1 and name2."""
if name1 == name2:
return 0
# Create the branches from the two desired nodes to the root
idx1 = self.name_index[name1]
branch1 = [(idx1, self.nodes[idx1].parent)]
idx2 = self.name_index[name2]
branch2 = [(idx2, self.nodes[idx2].parent)]
# Trace the path to the root for both nodes
while branch1[-1][1] != -1:
current_idx = branch1[-1][1]
branch1.append((current_idx, self.nodes[current_idx].parent))
while branch2[-1][1] != -1:
current_idx = branch2[-1][1]
branch2.append((current_idx, self.nodes[current_idx].parent))
# Calculate the distance as the sum of edge weights in the symmetric difference of paths
return sum([self.edge_weight[edge] for edge in set(branch1) ^ set(branch2)])
# Sample input
sample_input = """
(dog:42,cat:33);
cat dog
((dog:4,cat:3):74,robot:98,elephant:58);
dog elephant
"""
input_lines = sample_input.strip().split('\n')
# Compute distances between pairs of nodes in each tree
distance_list = []
for i in range(0, len(input_lines) - 1, 3):
tree = input_lines[i]
nodeA, nodeB = input_lines[i + 1].split()
distance_list.append(str(WeightedNewick(tree).distance(nodeA, nodeB)))
# Print the computed distances
print(" ".join(distance_list))63.4 Explanation
- Node Class:
- This clas represents a node in the tree.
- Each node has a number, a parent, and an optional name.
- The
__repr__method provides a string representation of the node.
- WeightedNewick Class:
- This clas constructs a tree from a Newick string and provides functionality to compute distances between nodes.
__init__: Initializes the tree, constructs it from the input data, and creates a mapping from node names to their indices.construct_tree: Parses the Newick string to build the tree structure and store edge weights.distance: Computes the distance between two nodes by tracing their paths to the root and summing the edge weights in the symmetric difference of these paths.
- Main Execution:
- The sample input is split into lines, and the trees and node pairs are extracted.
- For each tree and node pair, a
WeightedNewickobject is created, and the distance between the specified nodes is computed. - The distances are printed in the required format.
64 Wobble Bonding and RNA Secondary Structures
Given an RNA string \(s\), we will augment the bonding graph of s by adding basepair edges connecting all occurrences of ‘U’ to all occurrences of ‘G’ in order to represent possible wobble base pairs.
We say that a matching in the bonding graph for s is valid if it is noncrossing (to prevent pseudoknots) and has the property that a basepair edge in the matching cannot connect symbols \(s_j\) and \(s_k\) unles \(k≥j+4\) (to prevent nearby nucleotides from base pairing).
Given: An RNA string \(s\) (of length at most 200 bp).
Return: The total number of distinct valid matchings of basepair edges in the bonding graph of \(s\). Assume that wobble base pairing is allowed.
64.1 Sample Dataset
AUGCUAGUACGGAGCGAGUCUAGCGAGCGAUGUCGUGAGUACUAUAUAUGCGCAUAAGCCACGU64.2 Sample Output
28485021997742164.3 Solution
To solve the problem of counting distinct valid matchings of basepair edges in an RNA string considering wobble base pairing (G-U pairs) and noncrossing constraints, we can use a dynamic programming approach. Below is a detailed explanation and implementation in Python:
64.4 Approach
- Dynamic Programming (DP) Setup:
- Define a 2D DP table
dpwheredp[i][j]represents the number of valid matchings for the substring of the RNA sequence from indexito indexj. - Base case:
dp[i][i-1] = 1for allibecause an empty substring has one valid matching (the empty matching).
- Define a 2D DP table
- Recursive Relation:
- For each pair of indices
iandjsuch thati < j, we consider the possibility of the base at positionipairing with any valid base at positionkwherei < k <= jandk >= i + 4(to respect the distance constraint). - The RNA bases can pair if they form a valid pair:
A-U,U-A,C-G,G-C,G-U, orU-G. - The number of matchings for substring
s[i:j+1]is calculated by splitting it into the matchings betweens[i]ands[k]and recursively solving for the substringss[i+1:k-1]ands[k+1:j].
- For each pair of indices
- Iterative Calculation:
- Fill in the DP table iteratively, starting from smaller substrings and building up to the entire string.
64.5 Implementation
def count_valid_matchings(rna):
n = len(rna)
dp = [[0] * n for _ in range(n)]
def can_pair(b1, b2):
return (b1 == 'A' and b2 == 'U') or (b1 == 'U' and b2 == 'A') or \
(b1 == 'C' and b2 == 'G') or (b1 == 'G' and b2 == 'C') or \
(b1 == 'G' and b2 == 'U') or (b1 == 'U' and b2 == 'G')
for i in range(n):
dp[i][i] = 1 # A single base has one valid matching (itself)
for length in range(1, n + 1): # length is the length of the substring
for i in range(n - length):
j = i + length
dp[i][j] = dp[i+1][j] # Case where s[i] is not paired
for k in range(i + 4, j + 1):
if can_pair(rna[i], rna[k]):
if k == j:
dp[i][j] += dp[i+1][k-1]
else:
dp[i][j] += dp[i+1][k-1] * dp[k+1][j]
return dp[0][n-1]
# Sample Dataset
rna = "AUGCUAGUACGGAGCGAGUCUAGCGAGCGAUGUCGUGAGUACUAUAUAUGCGCAUAAGCCACGU"
# Output the result
print(count_valid_matchings(rna))64.6 Explanation
- Base Case:
- Each single base (or empty substring) has one valid matching (
dp[i][i] = 1).
- Each single base (or empty substring) has one valid matching (
- Filling DP Table:
- For each possible substring length, calculate the number of valid matchings by considering all possible pairs for the first base and ensuring the substrings formed by removing the matched bases also have valid matchings.
- Helper Function:
can_pairchecks if two bases can pair according to the given rules including wobble base pairing.
65 Counting Disease Carriers
To model the Hardy-Weinberg principle, assume that we have a population of \(N\) diploid individuals. If an allele is in genetic equilibrium, then because mating is random, we may view the \(2N\) chromosomes as receiving their alleles uniformly. In other words, if there are mm dominant alleles, then the probability of a selected chromosome exhibiting the dominant allele is simply \(p= \frac{m}{2N}\).
Because the first assumption of genetic equilibrium states that the population is so large as to be ignored, we will assume that \(N\) is infinite, so that we only need to concern ourselves with the value of \(p\).
Given: An array \(A\) for which \(A[k]\) represents the proportion of homozygous recessive individuals for the \(k\)-th Mendelian factor in a diploid population. Assume that the population is in genetic equilibrium for all factors.
Return: An array \(B\) having the same length as \(A\) in which \(B[k]\) represents the probability that a randomly selected individual carries at least one copy of the recessive allele for the \(k\)-th factor.
65.1 Sample Dataset
0.1 0.25 0.565.2 Sample Output
0.532 0.75 0.91465.3 Solution
To solve the problem of determining the probability that a randomly selected individual carries at least one copy of the recessive allele for each Mendelian factor, we need to work with the Hardy-Weinberg equilibrium principles.
65.4 Steps to Solve
- Given Data:
- Array \(A\) where \(A[k]\) is the proportion of homozygous recessive individuals for the \(k\)-th factor.
- We need to find an array \(B\) where \(B[k]\) is the probability that a randomly selected individual carries at least one copy of the recessive allele for the \(k\)-th factor.
- Hardy-Weinberg Principle:
- In genetic equilibrium, the proportion of homozygous recessive individuals (denoted \(q^2\)) is \(A[k]\).
- The recessive allele frequency \(q\) is the square root of \(A[k]\), i.e., \(q = \sqrt{A[k]}\).
- The dominant allele frequency \(p\) is \(1 - q\).
- Carrier Probability:
- The probability that an individual carries at least one recessive allele (i.e., they are either heterozygous or homozygous recessive) is given by \(1 - p^2\).
- Since \(p = 1 - q\), the carrier probability becomes \(1 - (1 - q)^2\).
- Calculation:
- For each \(k\): \[ q = \sqrt{A[k]} \] \[ B[k] = 1 - (1 - q)^2 \]
65.5 Implementation
Here’s how you can implement this in Python:
import math
def calculate_carrier_probability(A):
B = []
for q_squared in A:
q = math.sqrt(q_squared)
p = 1 - q
carrier_probability = 1 - p**2
B.append(carrier_probability)
return B
# Sample Dataset
sample_input = """
0.1 0.25 0.5"""
A = [float(x) for x in sample_input.strip().split()]
# Calculate the carrier probabilities
B = calculate_carrier_probability(A)
# Print the results formatted to three decimal places
print(" ".join(f"{prob:f}" for prob in B))65.6 Explanation
- Function Definition:
calculate_carrier_probabilitytakes the array \(A\) as input and returns the array \(B\).
- Loop Through \(A\):
- For each element in \(A\):
- Compute \(q\) as the square root of the element.
- Compute \(p\) as \(1 - q\).
- Compute the carrier probability using \(1 - p^2\).
- Append the result to \(B\).
- For each element in \(A\):
- Output:
- Format the output to three decimal places for better readability.
66 Creating a Character Table from Genetic Strings
A collection of strings is characterizable if there are at most two possible choices for the symbol at each position of the strings.
Given: A collection of at most 100 characterizable DNA strings, each of length at most 300 bp.
Return: A character table for which each nontrivial character encodes the symbol choice at a single position of the strings. (Note: the choice of assigning ‘1’ and ‘0’ to the two states of each SNP in the strings is arbitrary.)
66.1 Sample Dataset
ATGCTACC
CGTTTACC
ATTCGACC
AGTCTCCC
CGTCTATC66.2 Sample Output
10110
1010066.3 Solution
def char_table_from_strings(dna_list):
"""
Builds a character table from a given list of DNA strings.
Parameters:
dna_list (list of str): A list of DNA strings.
Returns:
set of str: A set containing nontrivial character rows.
"""
character_table = set()
# Iterate over each position in the DNA strings
for pos in range(len(dna_list[0])):
# Determine the reference character at the current position from the first DNA string
ref_char = dna_list[0][pos]
# Create a binary array indicating the presence of the reference character at the current position
char_array = [int(dna[pos] == ref_char) for dna in dna_list]
# Check if the character array is nontrivial (i.e., it has both 0s and 1s but not all 0s or all 1s)
if 1 < sum(char_array) < len(dna_list) - 1:
# Convert the binary array to a string and add it to the character table
character_table.add(''.join(map(str, char_array)))
return character_table
# Sample input
sample_input = """
ATGCTACC
CGTTTACC
ATTCGACC
AGTCTCCC
CGTCTATC
"""
# Convert the input string to a list of DNA strings
dna_list = sample_input.strip().split("\n")
# Get the character table
character_table = char_table_from_strings(dna_list)
# Print the character table
for row in character_table:
print(row)66.4 Explanation
Function Definition: The function
char_table_from_stringsis defined to take a list of DNA strings and return a set of nontrivial character rows.Initialize Character Table: An empty set
character_tableis initialized to store the nontrivial character rows.Iterate Over Positions: A loop iterates over each position in the DNA strings. The length of the first string is used to determine the number of positions.
Reference Character: For each position, the reference character
ref_charis taken from the first DNA string.Create Binary Array: A binary array
char_arrayis created using a list comprehension. For each DNA string, it checks if the character at the current position matches the reference character and records1if it does and0if it doesn’t.Check Nontrivial Condition: The array is considered nontrivial if it contains both
0sand1sbut is not all0sor all1s. This is checked using the condition1 < sum(char_array) < len(dna_list) - 1.Add to Character Table: If the binary array is nontrivial, it is converted to a string and added to the
character_tableset.Return Character Table: The function returns the
character_tableset containing all nontrivial character rows.Sample Input: The sample input is given as a multiline string, which is converted to a list of DNA strings by stripping and splitting by newline characters.
Generate and Print Character Table: The character table is generated by calling the function and printed row by row.
67 Counting Optimal Alignments
Recall from “Edit Distance Alignment” that if \(s′\) and \(t′\) are the augmented strings corresponding to an alignment of strings \(s\) and \(t\), then the edit alignment score of \(s′\) and \(t′\) was given by the Hamming distance \(dH(s′,t′)\) (because \(s′\) and \(t′\) have the same length and already include gap symbols to denote insertions/deletions).
As a result, we obtain \(d_E(s,t)=min_{s′,t′} dH(s′,t′)\), where the minimum is taken over all alignments of \(s\) and \(t\). Strings \(s′\) and \(t′\) achieving this minimum correspond to an optimal alignment with respect to edit alignment score.
Given: Two protein strings \(s\) and \(t\) in FASTA format, each of length at most 1000 aa.
Return: The total number of optimal alignments of \(s\) and \(t\) with respect to edit alignment score, modulo \(134,217,727 (2^{27}-1)\).
67.1 Sample Dataset
>Rosalind_78
PLEASANTLY
>Rosalind_33
MEANLY67.2 Sample Output
467.3 Solution
def parse_fasta(fasta_string):
'''Parses a FASTA format string and returns the sequences in a list.'''
sequences = []
sequence = []
for line in fasta_string.strip().split('\n'):
if line.startswith('>'):
if sequence:
sequences.append(''.join(sequence))
sequence = []
else:
sequence.append(line.strip())
if sequence:
sequences.append(''.join(sequence))
return sequences
def count_optimal_alignments(s, t):
'''Counts the total number of optimal alignments of s and t with respect to edit alignment score.'''
MOD = 134217727 # Modulo value to prevent overflow
m, n = len(s), len(t)
# Initialize DP tables
dp = [[0] * (n + 1) for _ in range(m + 1)] # Table for edit distances
count = [[0] * (n + 1) for _ in range(m + 1)] # Table for counting optimal alignments
# Base cases: edit distance and count for aligning to empty string
for i in range(m + 1):
dp[i][0] = i
count[i][0] = 1
for j in range(n + 1):
dp[0][j] = j
count[0][j] = 1
# Fill the DP tables
for i in range(1, m + 1):
for j in range(1, n + 1):
# Calculate the cost of insert, delete, and replace operations
insert_cost = dp[i][j-1] + 1
delete_cost = dp[i-1][j] + 1
replace_cost = dp[i-1][j-1] + (0 if s[i-1] == t[j-1] else 1)
# Find the minimum cost among the three operations
dp[i][j] = min(insert_cost, delete_cost, replace_cost)
# Count the number of ways to achieve this minimum cost
if dp[i][j] == insert_cost:
count[i][j] += count[i][j-1]
if dp[i][j] == delete_cost:
count[i][j] += count[i-1][j]
if dp[i][j] == replace_cost:
count[i][j] += count[i-1][j-1]
# Apply the modulo to keep the count manageable
count[i][j] %= MOD
return count[m][n]
# Sample dataset in FASTA format
sample_input = """
>Rosalind_78
PLEASANTLY
>Rosalind_33
MEANLY
"""
# Parse the FASTA input to get the sequences
sequences = parse_fasta(sample_input)
s, t = sequences[0], sequences[1]
# Get the number of optimal alignments
result = count_optimal_alignments(s, t)
# Print the result
print(result)67.4 Explanation
- FASTA Parsing (
parse_fasta):- This function reads a FASTA formatted string and extracts sequences.
- It initializes an empty list
sequencesto store the parsed sequences and another listsequenceto build each sequence. - It iterates over each line of the input string:
- If a line starts with
>, it indicates a new sequence header. Ifsequenceis not empty, it joins its elements into a single string and adds it tosequences. - If a line does not start with
>, it is part of a sequence, so it is added tosequence.
- If a line starts with
- After the loop, any remaining sequence is added to
sequences. - The function returns the list of sequences.
- Counting Optimal Alignments (
count_optimal_alignments):- This function calculates the number of optimal alignments between two strings
sandt. MODis a large prime number used to keep the counts within manageable limits.dpis a table wheredp[i][j]stores the minimum edit distance between the firsticharacters ofsand the firstjcharacters oft.countis a table wherecount[i][j]stores the number of optimal alignments that result in the minimum edit distance for the firsticharacters ofsand the firstjcharacters oft.- The base cases initialize the first row and first column of
dpandcountto represent alignments with an empty string. - The nested loops fill in the
dpandcounttables by considering insertion, deletion, and replacement operations. - The minimum cost operation is selected, and the number of ways to achieve this cost is counted.
- The result is the number of optimal alignments for the entire strings
sandt, stored incount[m][n].
- This function calculates the number of optimal alignments between two strings
- Main Execution:
- The sample input is given in FASTA format.
- The
parse_fastafunction is called to extract the sequences. - The
count_optimal_alignmentsfunction is called with the parsed sequences to get the number of optimal alignments. - The result is printed.
68 Counting Unrooted Binary Trees
Two unrooted binary trees \(T_1\) and \(T_2\) having the same n labeled leaves are considered to be equivalent if there is some assignment of labels to the internal nodes of \(T_1\) and \(T_2\) so that the adjacency lists of the two trees coincide. As a result, note that \(T_1\) and \(T_2\) must have the same splits; conversely, if the two trees do not have the same splits, then they are considered distinct.
Let \(b(n)\) denote the total number of distinct unrooted binary trees having n labeled leaves.
Given: A positive integer \(n\) (\(n≤1000\)).
Return: The value of \(b(n)\) modulo 1,000,000.
68.1 Sample Dataset
568.2 Sample Output
1568.3 Solution
import functools
def count_unrooted_binary_trees(num_leaves: int) -> int:
"""
Returns the number of unrooted binary trees with num_leaves leaves,
modulo 1,000,000.
"""
MODULO = 10**6
def double_factorial(n: int) -> int:
result = 1
for i in range(n, 1, -2):
result = (result * i) % MODULO
return result
return double_factorial(2 * num_leaves - 5)
# Sample input
sample_input = "5"
num_leaves = int(sample_input.strip())
# Get the number of unrooted binary trees
tree_count = count_unrooted_binary_trees(num_leaves)
# Print the result
print(tree_count)##$ Explanation
- Double Factorial Calculation without Lambda:
- The
double_factorialfunction is now implemented using a simpleforloop. - The loop iterates over the range from
ndown to 1, stepping by -2 (to get only odd numbers). - In each iteration, the current
resultis multiplied byiand taken modulo (10^6). - This avoids the use of a lambda function and
functools.reduce.
- The
- Rest of the Code:
- The rest of the code remains unchanged.
- The
count_unrooted_binary_treesfunction callsdouble_factorialwith2 * num_leaves - 5to compute the number of unrooted binary trees. - The result is printed after parsing the sample input.
69 Global Alignment with Scoring Matrix
To penalize symbol substitutions differently depending on which two symbols are involved in the substitution, we obtain a scoring matrix \(S\) in which \(S_{i,j}\) represents the (negative) score assigned to a substitution of the iith symbol of our alphabet \(𝒜\) with the \(j\)th symbol of \(𝒜\).
A gap penalty is the component deducted from alignment score due to the presence of a gap. A gap penalty may be a function of the length of the gap; for example, a linear gap penalty is a constant \(g\) such that each inserted or deleted symbol is charged \(g\); as a result, the cost of a gap of length \(L\) is equal to \(gL\).
Given: Two protein strings \(s\) and \(t\) in FASTA format (each of length at most 1000 aa).
Return: The maximum alignment score between \(s\) and \(t\). Use:
- The BLOSUM62 scoring matrix.
- Linear gap penalty equal to 5 (i.e., a cost of -5 is assessed for each gap symbol).
69.1 Sample Dataset
>Rosalind_67
PLEASANTLY
>Rosalind_17
MEANLY69.2 Sample Output
869.3 Solution
To solve the problem of finding the maximum alignment score between two protein strings using the BLOSUM62 scoring matrix and a linear gap penalty of 5, we need to implement the Needleman-Wunsch algorithm for global sequence alignment. This involves dynamic programming to compute the optimal alignment score.
Here is the step-by-step explanation of the solution along with the Python code implementation:
- Parse the FASTA input to extract the two protein sequences.
- Set up the BLOSUM62 scoring matrix.
- Implement the Needleman-Wunsch algorithm to compute the alignment score using the given scoring matrix and gap penalty.
def parse_fasta(fasta_string):
'''Parses a FASTA format string and returns the sequences in a list.'''
sequences = []
sequence = []
for line in fasta_string.strip().split('\n'):
if line.startswith('>'):
if sequence:
sequences.append(''.join(sequence))
sequence = []
else:
sequence.append(line.strip())
if sequence:
sequences.append(''.join(sequence))
return sequences
# BLOSUM62 matrix
blosum62_str = """
A C D E F G H I K L M N P Q R S T V W Y
A 4 0 -2 -1 -2 0 -2 -1 -1 -1 -1 -2 -1 -1 -1 1 0 0 -3 -2
C 0 9 -3 -4 -2 -3 -3 -1 -3 -1 -1 -3 -3 -3 -3 -1 -1 -1 -2 -2
D -2 -3 6 2 -3 -1 -1 -3 -1 -4 -3 1 -1 0 -2 0 -1 -3 -4 -3
E -1 -4 2 5 -3 -2 0 -3 1 -2 -2 0 -1 2 0 0 -1 -2 -3 -2
F -2 -2 -3 -3 6 -3 -1 0 -3 0 0 -3 -4 -3 -3 -2 -2 -1 1 3
G 0 -3 -1 -2 -3 6 -2 -4 -2 -4 -3 0 -2 -2 -3 0 -2 -3 -2 -3
H -2 -3 -1 0 -1 -2 8 -3 -1 -3 -2 1 -2 0 0 -1 -2 -3 -2 2
I -1 -1 -3 -3 0 -4 -3 4 -3 2 1 -3 -3 -3 -3 -2 -1 3 -3 -1
K -1 -3 -1 1 -3 -2 -1 -3 5 -2 -1 0 -1 1 2 0 -1 -2 -3 -2
L -1 -1 -4 -2 0 -4 -3 2 -2 4 2 -3 -3 -2 -2 -2 -1 1 -2 -1
M -1 -1 -3 -2 0 -3 -2 1 -1 2 5 -2 -2 0 -1 -1 -1 1 -1 -1
N -2 -3 1 0 -3 0 1 -3 0 -3 -2 6 -2 0 0 1 0 -3 -4 -2
P -1 -3 -1 -1 -4 -2 -2 -3 -1 -3 -2 -2 7 -1 -2 -1 -1 -3 -4 -3
Q -1 -3 0 2 -3 -2 0 -3 1 -2 0 0 -1 5 1 0 -1 -2 -2 -1
R -1 -3 -2 0 -3 -3 0 -3 2 -2 -1 0 -2 1 5 -1 -1 -3 -3 -2
S 1 -1 0 0 -2 0 -1 -2 0 -2 -1 1 -1 0 -1 4 1 -2 -3 -2
T 0 -1 -1 -1 -2 -2 -2 -1 -1 -1 -1 0 -1 -1 -1 1 5 0 -2 -2
V 0 -1 -3 -2 -1 -3 -3 3 -2 1 1 -3 -3 -2 -3 -2 0 4 -3 -1
W -3 -2 -4 -3 1 -2 -2 -3 -3 -2 -1 -4 -4 -2 -3 -3 -2 -3 11 2
Y -2 -2 -3 -2 3 -3 2 -1 -2 -1 -1 -2 -3 -1 -2 -2 -2 -1 2 7
"""
def parse_blosum62(matrix_str):
"""Parse the BLOSUM62 matrix from a string."""
lines = matrix_str.strip().split('\n')
headers = lines[0].split()
matrix = {}
for line in lines[1:]:
values = line.split()
row = values[0]
scores = list(map(int, values[1:]))
for col, score in zip(headers, scores):
matrix[(row, col)] = score
return matrix
def needleman_wunsch(s, t, blosum62, gap_penalty):
"""Perform the Needleman-Wunsch algorithm for global alignment."""
m, n = len(s), len(t)
dp = [[0] * (n + 1) for _ in range(m + 1)]
# Initialize dp table with gap penalties
for i in range(1, m + 1):
dp[i][0] = dp[i - 1][0] + gap_penalty
for j in range(1, n + 1):
dp[0][j] = dp[0][j - 1] + gap_penalty
# Fill the dp table
for i in range(1, m + 1):
for j in range(1, n + 1):
match = dp[i - 1][j - 1] + blosum62[(s[i - 1], t[j - 1])]
delete = dp[i - 1][j] + gap_penalty
insert = dp[i][j - 1] + gap_penalty
dp[i][j] = max(match, delete, insert)
return dp[m][n]
# Sample dataset in FASTA format
sample_input = """
>Rosalind_67
PLEASANTLY
>Rosalind_17
MEANLY
"""
# Parse the FASTA input to get the sequences
sequences = parse_fasta(sample_input)
s, t = sequences[0], sequences[1]
# Parse the BLOSUM62 matrix
blosum62 = parse_blosum62(blosum62_str)
# Set the gap penalty
gap_penalty = -5
# Get the maximum alignment score using Needleman-Wunsch algorithm
result = needleman_wunsch(s, t, blosum62, gap_penalty)
# Print the result
print(result)69.4 Explanation of the Code
- Parsing FASTA Input:
parse_fastafunction reads the input in FASTA format and returns the sequences in a list.
- BLOSUM62 Scoring Matrix:
parse_blosum62function parses the BLOSUM62 matrix string and stores the scores in a dictionary for easy lookup.
- Needleman-Wunsch Algorithm:
needleman_wunschfunction implements the dynamic programming algorithm to compute the global alignment score.- The
dptable is initialized with gap penalties. - The table is filled based on the scores for matches, insertions, and deletions.
- The final alignment score is found in
dp[m][n].
- Execution:
- The sequences are parsed, the scoring matrix is loaded, and the alignment score is computed using the Needleman-Wunsch algorithm.
- The result is printed as the maximum alignment score.
This implementation ensures the alignment score is computed efficiently even for long protein sequences, utilizing the scoring matrix and gap penalties correctly.
70 Genome Assembly with Perfect Coverage
A circular string is a string that does not have an initial or terminal element; instead, the string is viewed as a necklace of symbols. We can represent a circular string as a string enclosed in parentheses. For example, consider the circular DNA string (ACGTAC), and note that because the string “wraps around” at the end, this circular string can equally be represented by (CGTACA), (GTACAC), (TACACG), (ACACGT), and (CACGTA). The definitions of substrings and superstrings are easy to generalize to the case of circular strings (keeping in mind that substrings are allowed to wrap around).
Given: A collection of (error-free) DNA \(k\)-mers (\(k≤50\)) taken from the same strand of a circular chromosome. In this dataset, all \(k\)-mers from this strand of the chromosome are present, and their de Bruijn graph consists of exactly one simple cycle.
Return: A cyclic superstring of minimal length containing the reads (thus corresponding to a candidate cyclic chromosome).
70.1 Sample Dataset
ATTAC
TACAG
GATTA
ACAGA
CAGAT
TTACA
AGATT70.2 Sample Output
ATTACAG70.3 Solution
def generate_coverings(current_string, edges, k):
"""
Generate all possible complete cycle coverings from the given edges.
Args:
- current_string: The current string being formed as part of the cycle.
- edges: Remaining edges in the De Bruijn graph to be used.
- k: Length of the k-mers.
Returns:
- A list of possible cycle coverings as strings.
"""
# Find the indices of edges that can be added next based on the current string.
next_edges_indices = [i for i, edge in enumerate(edges) if edge[0] == current_string[-k+1:]]
# If no more edges can be added:
if not next_edges_indices:
# Return the current string if all edges have been used (perfect covering).
return [current_string] if not edges else []
# Otherwise, recursively generate coverings with each possible next edge.
possible_coverings = []
for i in next_edges_indices:
next_string = current_string + edges[i][1][-1]
remaining_edges = edges[:i] + edges[i+1:]
possible_coverings.append(generate_coverings(next_string, remaining_edges, k))
return possible_coverings
def flatten(nested_list):
"""
Flattens a nested list into a single list.
Args:
- nested_list: A list that may contain other nested lists.
Yields:
- Individual elements from the nested list, flattened.
"""
for item in nested_list:
if isinstance(item, list):
yield from flatten(item)
else:
yield item
# Sample input data
sample_input = """
ATTAC
TACAG
GATTA
ACAGA
CAGAT
TTACA
AGATT"""
# Split input into k-mers
k_mers = sample_input.strip().split("\n")
# Create edges of the De Bruijn graph from the k-mers
k = len(k_mers[0])
create_edge = lambda k_mer: [k_mer[:k-1], k_mer[1:]]
de_bruijn_edges = [create_edge(k_mer) for k_mer in k_mers[1:]]
# Generate all possible circular strings
circular_strings = set(flatten(generate_coverings(k_mers[0], de_bruijn_edges, k)))
# Trim each circular string to the appropriate length (number of k-mers)
circular_strings = [cycle[:len(k_mers)] for cycle in circular_strings]
# Print the resulting circular strings
print('\n'.join(circular_strings))70.4 Explanation of the Code
- generate_coverings Function:
- Purpose: This function recursively generates all possible cycle coverings (i.e., circular strings) by extending the current string with valid edges from the De Bruijn graph.
- How it works:
- It looks for edges that can be appended to the current string (based on the last \(k-1\) characters of the string).
- If no valid edges are left, it checks if all edges have been used (indicating a perfect covering). If so, it returns the current string.
- If there are valid edges, it recursively tries to extend the string with each possible edge and collects all possible coverings.
- flatten Function:
- Purpose: This helper function is used to flatten a nested list into a single-level list.
- How it works: It recursively traverses the nested list and yields individual elements, effectively flattening the list.
- Main Execution:
- k_mers: The input strings are split into individual \(k\)-mers.
- de_bruijn_edges: This creates the edges of the De Bruijn graph. Each \(k\)-mer is split into its prefix and suffix of length \(k-1\).
- generate_coverings: This function is called with the first \(k\)-mer as the starting point, and it generates all possible circular strings by finding all Eulerian cycles in the graph.
- circular_strings: The resulting strings are then trimmed to the length of the input (number of \(k\)-mers) to ensure that only the desired cyclic superstrings are printed.
71 Matching a Spectrum to a Protein
The complete spectrum of a weighted string \(s\) is the multiset \(S[s]\) containing the weights of every prefix and suffix of \(s\).
Given: A positive integer \(n\) followed by a collection of \(n\) protein strings \(s_1\), \(s_2\), \(...\), \(s_n\) and a multiset \(R\) of positive numbers (corresponding to the complete spectrum of some unknown protein string).
Return: The maximum multiplicity of \(R⊖S[s_k]\) taken over all strings \(s_k\), followed by the string \(s_k\) for which this maximum multiplicity occurs (you may output any such value if multiple solutions exist).
71.1 Sample Dataset
4
GSDMQS
VWICN
IASWMQS
PVSMGAD
445.17838
115.02694
186.07931
314.13789
317.1198
215.0906171.2 Sample Output
3
IASWMQS71.3 Solution
from decimal import Decimal
from collections import defaultdict
def get_protein_weights():
"""Returns a dictionary mapping amino acids to their weights."""
return {
'G': Decimal('57.02146'), 'A': Decimal('71.03711'), 'S': Decimal('87.03203'),
'P': Decimal('97.05276'), 'V': Decimal('99.06841'), 'T': Decimal('101.04768'),
'C': Decimal('103.00919'), 'I': Decimal('113.08406'), 'L': Decimal('113.08406'),
'N': Decimal('114.04293'), 'D': Decimal('115.02694'), 'Q': Decimal('128.05858'),
'K': Decimal('128.09496'), 'E': Decimal('129.04259'), 'M': Decimal('131.04049'),
'H': Decimal('137.05891'), 'F': Decimal('147.06841'), 'R': Decimal('156.10111'),
'Y': Decimal('163.06333'), 'W': Decimal('186.07931')
}
def calculate_weight(protein_sequence):
"""Calculates the total weight of a given protein sequence based on amino acid weights."""
weights = get_protein_weights()
total_weight = Decimal('0.0')
for amino_acid in protein_sequence:
total_weight += weights[amino_acid]
return total_weight
def calculate_multiplicity(proteins, spectrum_weights):
"""
Calculates the maximum multiplicity of spectrum weights for each protein
and identifies the protein with the highest multiplicity.
"""
max_multiplicity = -1
best_protein = None
for protein in proteins:
# Compute the spectrum weights for all prefixes and suffixes of the protein
spectrum = []
for i in range(1, len(protein) + 1):
spectrum.append(calculate_weight(protein[:i]))
for i in range(len(protein)):
spectrum.append(calculate_weight(protein[i:]))
spectrum_weights_count = defaultdict(int)
# Count the differences between the spectrum weights and given weights
for protein_weight in spectrum:
for given_weight in spectrum_weights:
diff = round(protein_weight - given_weight, 3)
spectrum_weights_count[diff] += 1
# Determine the maximum multiplicity for the current protein
current_multiplicity = max(spectrum_weights_count.values(), default=0)
if current_multiplicity > max_multiplicity:
max_multiplicity = current_multiplicity
best_protein = protein
return max_multiplicity, best_protein
# Sample input (for demonstration purposes)
sample_input = """
4
GSDMQS
VWICN
IASWMQS
PVSMGAD
445.17838
115.02694
186.07931
314.13789
317.1198
215.09061
"""
# Parse the input
lines = sample_input.strip().split("\n")
number_of_proteins = int(lines[0])
protein_sequences = lines[1:number_of_proteins + 1]
spectrum_weights = sorted(map(Decimal, lines[number_of_proteins + 1:]))
# Calculate the maximum multiplicity and the corresponding protein
max_multiplicity, best_protein = calculate_multiplicity(protein_sequences, spectrum_weights)
# Print the results
print(max_multiplicity)
print(best_protein)71.4 Explanation of the Code
get_protein_weights(): Returns a dictionary mapping each amino acid to its corresponding weight using theDecimaltype for precision.calculate_weight(protein_sequence): Computes the total weight of a protein sequence by summing the weights of its amino acids.calculate_multiplicity(proteins, spectrum_weights):- Iterates through each protein sequence to compute the weights of all possible prefixes and suffixes.
- Uses a
defaultdictto count how often the difference between each protein weight and given spectrum weight appears. - Finds and returns the protein with the highest multiplicity of such differences.
Input Parsing:
- Reads and parses the sample input to extract the number of proteins, the list of protein sequences, and the list of spectrum weights.
Results:
- Calls
calculate_multiplicityto get the protein with the maximum multiplicity and prints the result.
- Calls
72 Quartets
A partial split of a set \(S\) of \(n\) taxa models a partial character and is denoted by \(A∣B\), where \(A\) and \(B\) are still the two disjoint subsets of taxa divided by the character. Unlike in the case of splits, we do not necessarily require that \(A∪B=S\); \((A∪B)c\) corresponds to those taxa for which we lack conclusive evidence regarding the character.
We can assemble a collection of partial characters into a generalized partial character table \(C\) in which the symbol \(x\) is placed in \(C_{i,j}\) if we do not have conclusive evidence regarding the \(j\)th taxon with respect to the \(i\)th partial character.
A quartet is a partial split \(A∣B\) in which both \(A\) and \(B\) contain precisely two elements. For the sake of simplicity, we often will consider quartets instead of partial characters. We say that a quartet \(A∣B\) is inferred from a partial split \(C∣D\) if \(A⊆C\) and \(B⊆D\) (or equivalently \(A⊆D\) and \(B⊆C\). For example, \({1,3}∣{2,4}\) and \({3,5}∣{2,4}\) can be inferred from \({1,3,5}∣{2,4}\).
Given: A partial character table \(C\).
Return: The collection of all quartets that can be inferred from the splits corresponding to the underlying characters of \(C\).
72.1 Sample Dataset
cat dog elephant ostrich mouse rabbit robot
01xxx00
x11xx00
111x00x72.2 Sample Output
{cat, dog} {mouse, rabbit}
{dog, elephant} {rabbit, robot}
{cat, elephant} {mouse, rabbit}
{dog, elephant} {mouse, rabbit}72.3 Solution
# Sample input data
data = """
cat dog elephant ostrich mouse rabbit robot
01xxx00
x11xx00
111x00x
"""
# Split the input data into lines
lines = data.strip().split("\n")
# Extract taxa (species) from the first line
taxa = lines[0].strip().split(' ')
# Initialize a set to store unique quartets
unique_quartets = set()
# Proces each line of the partial character table
for line in lines[1:]:
# Initialize lists to hold taxa for two groups
group_C = []
group_D = []
# Classify taxa based on the partial character table
for i in range(len(line)):
if line[i] == '1':
group_C.append(taxa[i])
elif line[i] == '0':
group_D.append(taxa[i])
# Ensure each group has at least two taxa to form a quartet
if len(group_C) >= 2 and len(group_D) >= 2:
# Generate all possible pairs for group C and group D
for i in range(len(group_C) - 1):
for j in range(i + 1, len(group_C)):
for k in range(len(group_D) - 1):
for l in range(k + 1, len(group_D)):
# Form pairs (A, B) from group_C and (C, D) from group_D
pair_A = tuple(sorted([group_C[i], group_C[j]]))
pair_B = tuple(sorted([group_D[k], group_D[l]]))
# Add the sorted quartet to the set
sorted_quartet = (pair_A, pair_B) if pair_A < pair_B else (pair_B, pair_A)
unique_quartets.add(sorted_quartet)
# Print each unique quartet in the required format
for quartet in unique_quartets:
pair_A, pair_B = quartet
print('{{{}, {}}} {{{}, {}}}'.format(pair_A[0], pair_A[1], pair_B[0], pair_B[1]))72.4 Explanation of the Code
- Input Data Handling:
data.strip().split("\n"): Split the input data into lines. The first line contains taxa names, and the subsequent lines contain the partial character table.
- Extracting Taxa:
taxa = lines[0].strip().split(' '): The first line is split into individual taxa names.
- Initialize Set for Quartets:
unique_quartets = set(): This set will store unique quartets to avoid duplicates.
- Processing Each Partial Character Table Line:
- For each line after the first one, initialize
group_Candgroup_Dto store taxa based on the partial character table values (1and0respectively). - Populate
group_Candgroup_Dbased on whether the character is1or0.
- For each line after the first one, initialize
- Forming Quartets:
- Ensure each group has at least two taxa to form pairs.
- Generate all possible pairs from
group_Candgroup_D. - Sort pairs and add them to the
unique_quartetsset, ensuring that each quartet is stored in a canonical (sorted) form to avoid duplicates.
- Output Results:
- For each unique quartet, format and print the result.
73 Using the Spectrum Graph to Infer Peptides
For a weighted alphabet \(𝒜\) and a collection \(L\) of positive real numbers, the spectrum graph of \(L\) is a digraph constructed in the following way. First, create a node for every real number in \(L\). Then, connect a pair of nodes with a directed edge \((u,v)\) if \(v>u\) and \(v−u\) is equal to the weight of a single symbol in \(𝒜\). We may then label the edge with this symbol.
In this problem, we say that a weighted string \(s=s_1s_2⋯s_n\) matches \(L\) if there is some increasing sequence of positive real numbers \((w1,w2,…,wn+1)\) in \(L\) such that \(w(s1)=w2−w1\), \(w(s2)=w3−w2\), …, and \(w(sn)=wn+1−wn\).
Given: A list \(L\) (of length at most 100) containing positive real numbers.
Return: The longest protein string that matches the spectrum graph of \(L\) (if multiple solutions exist, you may output any one of them). Consult the monoisotopic mas table.
73.1 Sample Dataset
3524.8542
3623.5245
3710.9335
3841.974
3929.00603
3970.0326
4026.05879
4057.0646
4083.0802573.2 Sample Output
SPG73.3 Solution
from collections import defaultdict
# Monoisotopic mas table for amino acids
mass_table = {
'A': 71.03711, 'C': 103.00919, 'D': 115.02694, 'E': 129.04259,
'F': 147.06841, 'G': 57.02146, 'H': 137.05891, 'I': 113.08406,
'K': 128.09496, 'L': 113.08406, 'M': 131.04049, 'N': 114.04293,
'P': 97.05276, 'Q': 128.05858, 'R': 156.10111, 'S': 87.03203,
'T': 101.04768, 'V': 99.06841, 'W': 186.07931, 'Y': 163.06333
}
def build_spectrum_graph(L, mass_table):
"""
Create a graph where each node represents a number in L.
Add a directed edge from u to v if v > u and the weight difference
matches any amino acid's mas in the mass_table.
"""
graph = defaultdict(list)
# Iterate over each pair of nodes (u, v) where v > u
for i, u in enumerate(L):
for j, v in enumerate(L):
if v > u:
# Calculate the weight difference between v and u
weight_diff = v - u
# Check if this weight difference matches any amino acid mass
for symbol, mas in mass_table.items():
if abs(weight_diff - mass) < 1e-5:
graph[u].append((v, symbol))
return graph
def find_longest_path(graph, start):
"""
Use depth-first search to find the longest path in the graph starting from 'start'.
"""
stack = [(start, '')] # Stack for DFS: (current_node, path_string)
longest_path = ''
# Store the longest path ending at each node
path_map = defaultdict(str)
while stack:
node, path = stack.pop()
# Update the longest path for the current node
if len(path) > len(path_map[node]):
path_map[node] = path
# Traverse neighbors
for neighbor, symbol in graph[node]:
stack.append((neighbor, path + symbol))
# Return the longest path found
longest_path = max(path_map.values(), key=len)
return longest_path
def find_longest_protein_string(L, mass_table):
"""
Build the spectrum graph and find the longest protein string.
"""
# Create the spectrum graph from the list L
graph = build_spectrum_graph(L, mass_table)
# Find the longest path starting from each node in L
longest_protein = ''
for node in L:
current_protein = find_longest_path(graph, node)
if len(current_protein) > len(longest_protein):
longest_protein = current_protein
return longest_protein
# Sample input
sample_input = """
3524.8542
3623.5245
3710.9335
3841.974
3929.00603
3970.0326
4026.05879
4057.0646
4083.08025"""
# Parse the sample input into a list of floats
L = [float(x) for x in sample_input.strip().split("\n")]
# Find and print the longest protein string
longest_protein = find_longest_protein_string(L, mass_table)
print(longest_protein)73.4 Explanation
- Monoisotopic Mas Table:
mass_tablemaps each amino acid to its mass.
- Graph Construction (
build_spectrum_graph):- Purpose: Create a directed graph where each node is a number from \(L\), and edges are added if the difference between nodes corresponds to the mas of an amino acid.
- Process:
- Iterate through each pair of numbers in \(L\) where the second number is greater than the first.
- Calculate the weight difference and check if it matches any mas in the
mass_table. - Add a directed edge between these nodes labeled with the corresponding amino acid.
- Finding the Longest Path (
find_longest_path):- Purpose: Determine the longest path in the graph starting from a given node using depth-first search (DFS).
- Process:
- Use a stack to explore nodes.
- Track the longest path ending at each node.
- Update the longest path found during traversal.
- Main Function (
find_longest_protein_string):- Purpose: Integrates the graph construction and longest path finding to return the longest protein string.
- Process:
- Build the spectrum graph.
- For each node, find the longest path starting from that node.
- Return the longest path found.
- Execution:
- Sample Input: Represents a list of mas values.
- Processing: Converts the sample input into a list of floats, finds the longest protein string, and prints it.
74 Encoding Suffix Trees
Given a string s having length \(n\), recall that its suffix tree \(T(s)\) is defined by the following properties:
- \(T(s)\) is a rooted tree having exactly n leaves.
- Every edge of \(T(s)\) is labeled with a substring of \(s∗\), where \(s∗\) is the string formed by adding a placeholder symbol
$to the end of \(s\). - Every internal node of \(T(s)\) other than the root has at least two children; i.e., it has degree at least 3.
- The substring labels for the edges leading down from a node to its children must begin with different symbols.
- By concatenating the substrings along edges, each path from the root to a leaf corresponds to a unique suffix of \(s∗\).
Given: A DNA string s of length at most 1kbp.
Return: The substrings of \(s∗\) encoding the edges of the suffix tree for \(s\). You may list these substrings in any order.
74.1 Sample Dataset
ATAAATG$74.2 Sample Output
A
A
ATG$
TG$
T
AAATG$
G$
T
AAATG$
G$
G$
$74.3 Solution
from collections import defaultdict
clas SuffixTree:
"""Creates a suffix tree for the provided word."""
def __init__(self, word):
"""Initializes the suffix tree."""
self.nodes = [self.Node(None, 0)] # Initialize with root node.
self.edges = dict() # Dictionary to store edges.
self.descendants_count = dict() # Cache for the number of descendants of nodes.
if isinstance(word, str): # Check if the input is a string.
self._build_suffix_tree(word)
clas Node:
"""Represents a node in the suffix tree."""
def __init__(self, parent, node_id):
self.parent = parent
self.node_id = node_id
self.children = []
def add_child(self, child_node):
self.children.append(child_node)
def remove_child(self, child_node):
self.children.remove(child_node)
def update_parent(self, new_parent):
self.parent = new_parent
def _build_suffix_tree(self, word):
"""Builds the suffix tree by adding each suffix of the word."""
if word[-1] != '$':
word += '$' # Ensure the word ends with the terminal symbol '$'.
self.word = word
self.length = len(self.word)
for i in range(self.length):
parent_node, edge_start, has_overlap = self._find_insertion_point(i, self.nodes[0])
if has_overlap:
existing_start, existing_end = self.edges[(parent_node.parent.node_id, parent_node.node_id)]
# Determine the length of the overlap.
overlap_length = 0
while self.word[edge_start:edge_start + overlap_length] == self.word[existing_start:existing_start + overlap_length]:
overlap_length += 1
# Create a new internal node at the point of insertion.
new_internal_node = self.Node(parent_node.parent, len(self.nodes))
new_internal_node.add_child(parent_node)
self._add_edge_and_node(parent_node.parent, existing_start, existing_start + overlap_length - 1, new_internal_node)
# Update the edge and parent relationship for the original child node.
del self.edges[(parent_node.parent.node_id, parent_node.node_id)]
parent_node.parent.remove_child(parent_node)
parent_node.update_parent(new_internal_node)
self.edges[(new_internal_node.node_id, parent_node.node_id)] = [existing_start + overlap_length - 1, existing_end]
# Add the remaining suffix as a new child node.
self._add_edge_and_node(new_internal_node, edge_start + overlap_length - 1, self.length)
else:
# No overlap, simply add the entire suffix as a new edge.
self._add_edge_and_node(parent_node, edge_start, self.length)
def _find_insertion_point(self, start_index, parent_node):
"""Determines where to insert a suffix into the tree."""
for child_node in parent_node.children:
edge_start, edge_end = self.edges[(parent_node.node_id, child_node.node_id)]
if self.word[start_index:start_index + edge_end - edge_start] == self.word[edge_start:edge_end]:
return self._find_insertion_point(start_index + edge_end - edge_start, child_node)
elif self.word[edge_start] == self.word[start_index]:
return child_node, start_index, True
return parent_node, start_index, False
def _add_edge_and_node(self, parent_node, edge_start, edge_end, child_node=None):
"""Adds a node and the corresponding edge to the suffix tree."""
if child_node is None:
child_node = self.Node(parent_node, len(self.nodes))
self.nodes.append(child_node)
parent_node.add_child(child_node)
self.edges[(parent_node.node_id, child_node.node_id)] = [edge_start, edge_end]
def get_edge_labels(self):
"""Returns the substrings representing the edges of the suffix tree."""
return [self.word[start:end] for start, end in self.edges.values()]
def count_total_descendants(self, node):
"""Calculates the total number of descendants of a given node."""
if node not in self.descendants_count:
self.descendants_count[node] = len(node.children) + sum(self.count_total_descendants(child) for child in node.children)
return self.descendants_count[node]
def get_node_label(self, node):
"""Returns the string represented by the path from the root to a given node."""
label = ''
while node.node_id != 0:
start, end = self.edges[(node.parent.node_id, node.node_id)]
label = self.word[start:end] + label
node = node.parent
return label.strip('$')
# Example usage
sample_input = "ATAAATG$"
suffix_tree = SuffixTree(sample_input)
result = '\n'.join(suffix_tree.get_edge_labels())
print(result)74.4 Explanation of the Code
- Clas Structure:
SuffixTreeclass: Manages the construction and representation of the suffix tree.Nodeclas (nested withinSuffixTree): Represents each node in the suffix tree. Each node has a parent, a unique identifier (node_id), and a list of child nodes.
- Initialization:
- The
SuffixTreeis initialized with a root node (withnode_id = 0). - The
_build_suffix_treemethod ensures the input word ends with the terminal symbol$and then processes each suffix of the word to build the tree.
- The
- Building the Tree:
- For each suffix,
_find_insertion_pointis called to determine where in the tree the suffix should be inserted. - If there’s an overlap with an existing edge, a new internal node is created, and the tree is split at the point of overlap.
- Otherwise, a new edge representing the suffix is added directly.
- For each suffix,
- Edge and Node Management:
- The
_add_edge_and_nodemethod handles the actual insertion of edges and nodes into the tree structure. - The
edgesdictionary maps parent-child relationships to the start and end indices of the corresponding substring in the word.
- The
- Retrieving Results:
get_edge_labels: Returns the list of substrings corresponding to all edges in the tree.count_total_descendants: Computes the number of descendants for any given node in the tree (cached to optimize repeated queries).get_node_label: Recovers the substring represented by a path from the root to a specific node.
75 Character-Based Phylogeny
Because a tree having \(n\) nodes has \(n−1\) edges (see “Completing a Tree”), removing a single edge from a tree will produce two smaller, disjoint trees. Recall from “Creating a Character Table” that for this reason, each edge of an unrooted binary tree corresponds to a split \(S∣S^c\), where \(S\) is a subset of the taxa.
A consistent character table is one whose characters’ splits do not conflict with the edge splits of some unrooted binary tree \(T\) on the n taxa. More precisely, \(S_1∣S^c_1\) conflicts with \(S_2∣S^c_2\) if all four intersections \(S_1∩S_2\), \(S_1∩S^c_2\), \(S^c_1∩S_2\), and \(S^c_1∩S^c_2\) are nonempty. As a simple example, consider the conflicting splits \(\{a,b\}∣\{c,d\}\) and \(\{a,c\}∣\{b,d\}\).
More generally, given a consistent character table \(C\), an unrooted binary tree \(T\) “models” \(C\) if the edge splits of \(T\) agree with the splits induced from the characters of \(C\).
Given: A list of n species (\(n≤80\)) and an \(n\)-column character table \(C\) in which the \(j\)th column denotes the \(j\)th species.
Return: An unrooted binary tree in Newick format that models \(C\).
75.1 Sample Dataset
cat dog elephant mouse rabbit rat
011101
001101
00110075.2 Sample Output
(((cat,rabbit),dog),(elephant,mouse),rat);75.3 Solution
from Bio import Phylo
import sys
def find_columns_to_unify(splits):
"""Finds two columns in the split matrix that should be unified into a single clade."""
for split in splits:
sum_split = sum(split)
if sum_split == 2:
# Find the pair of columns where the sum is exactly 2.
return tuple(i for i, value in enumerate(split) if value == 1)
elif sum_split == len(split) - 2:
# Find the pair of columns where the sum is all but 2.
return tuple(i for i, value in enumerate(split) if value == 0)
raise ValueError('No columns to unify found!', splits)
def print_clade_trees(clades):
"""Prints the clades as Newick formatted trees."""
for clade in clades:
tree = Phylo.BaseTree.Tree.from_clade(clade)
Phylo.write(tree, sys.stdout, 'newick', plain=True)
def build_phylogenetic_tree(input_lines):
"""Builds a phylogenetic tree based on the input split matrix."""
clades = [Phylo.BaseTree.Clade(name=name) for name in input_lines[0].split()]
splits = []
for line in input_lines[1:]:
splits.append([int(x) for x in line])
while splits:
col1, col2 = find_columns_to_unify(splits)
# Remove the second of the unified columns from the splits
for split in splits:
split.pop(col2)
# Remove trivial splits where all entries are 1 or all but one are 1
splits = [split for split in splits if 1 < sum(split) < len(split) - 1]
# Unify the clades corresponding to the selected columns
clades[col1] = Phylo.BaseTree.Clade(clades=[clades[col1], clades[col2]])
clades.pop(col2)
# Final clade to represent the complete phylogenetic tree
final_clade = Phylo.BaseTree.Clade(clades=clades)
print_clade_trees([final_clade])
# Sample Input
sample_input = """
cat dog elephant mouse rabbit rat
011101
001101
001100
"""
input_lines = sample_input.strip().split("\n")
build_phylogenetic_tree(input_lines)75.4 Explanation of the Code
- Purpose:
- The code is designed to build a phylogenetic tree based on a set of species (or objects) and their binary split representations. The input consists of species names followed by rows of binary digits representing splits between species.
- Functions:
find_columns_to_unify(splits):- This function identifies two columns in the split matrix that can be unified into a single clade.
- It looks for a pair of columns where the sum of the values in the columns equals 2 (indicating that exactly two species share a common clade) or equals the number of species minus 2 (indicating all but two species share a clade).
print_clade_trees(clades):- This function prints the clades in Newick format, which is a common format for representing phylogenetic trees.
build_phylogenetic_tree(input_lines):- This is the main function that constructs the phylogenetic tree.
- It first parses the input to create a list of clades, one for each species.
- Then it iterates through the split matrix, repeatedly unifying clades based on the identified columns until only one clade (the final tree) remains.
- Process:
- Input Parsing:
- The input string is split into lines, with the first line containing species names and the subsequent lines containing binary splits.
- Tree Construction:
- The code processes the splits by repeatedly identifying pairs of species to unify (using the
find_columns_to_unifyfunction). - After unifying species into clades, it modifies the split matrix by removing the unified columns and trivial splits.
- This continues until only one clade remains, representing the complete phylogenetic tree.
- The code processes the splits by repeatedly identifying pairs of species to unify (using the
- Input Parsing:
- Output:
- The resulting phylogenetic tree is printed in Newick format, which can be visualized or further analyzed using tools that support this format.
76 Counting Quartets
A quartet \(AB∣CD\) is consistent with a binary tree \(T\) if the quartet can be inferred from one of the splits of \(T\) (see “Quartets” for a description of inferring quartets from splits).
Let \(q(T)\) denote the total number of quartets that are consistent with \(T\).
Given: A positive integer n (\(4≤n≤5000\)), followed by an unrooted binary tree \(T\) in Newick format on \(n\) taxa.
Return: The value of \(q(T)\) modulo 1,000,000.
76.1 Sample Dataset
6
(lobster,(cat,dog),(caterpillar,(elephant,mouse)));76.2 Sample Output
1576.3 Solution
# Sample Input
sample_input = """
6
(lobster,(cat,dog),(caterpillar,(elephant,mouse)));
"""
# Parse input lines
input_lines = sample_input.strip().split("\n")
n = int(input_lines[0])
# Initialize a memoization list to store factorials
factorials = [1] * (n + 1) # Initialize with 1 for factorial(0)
# Calculate all factorials from 1 to n and store in the list
for i in range(1, n + 1):
factorials[i] = i * factorials[i - 1]
# Calculate the number of possible quartets using the combination formula
# C(n, 4) = n! / ((n-4)! * 4!)
num_quartets = (factorials[n] // (factorials[n - 4] * factorials[4])) % 1000000
# Output the result
print(num_quartets)76.4 Explanation
- Input Parsing:
- The input is a string representing the number of taxa
nand a Newick formatted tree. The first line is split to extractn.
- The input is a string representing the number of taxa
- Factorial Calculation:
- We need to calculate the number of quartets possible, which involves computing combinations. The combination formula
C(n, 4)is used to calculate how many ways we can choose 4 taxa fromn, and it requires calculating factorials. - We initialize a list
factorialswithn+1elements, all set to1. This list will store factorial values from0!ton!. - A loop is used to calculate each factorial iteratively and store it in the list.
- We need to calculate the number of quartets possible, which involves computing combinations. The combination formula
- Combination Calculation:
- The number of quartets is calculated using the formula
C(n, 4) = n! / ((n-4)! * 4!). - The combination result is then taken modulo
1,000,000to meet the problem’s requirement.
- The number of quartets is calculated using the formula
- Output:
- The final result is printed.
76.5 Key Concepts
- Factorials: Factorials are calculated iteratively and stored in a list to avoid recalculating the same value multiple times.
- Combinations: The formula for combinations
C(n, 4)is used to find out how many quartets can be formed fromntaxa. - Modulo Operation: Since the number of quartets can be large, the result is taken modulo
1,000,000.
77 Enumerating Unrooted Binary Trees
Recall the definition of Newick format from “Distances in Trees” as a way of encoding trees.
Given: A collection of species names representing \(n\) taxa.
Return: A list containing all unrooted binary trees whose leaves are these \(n\) taxa. Trees should be given in Newick format, with one tree on each line; the order of the trees is unimportant.
77.1 Sample Dataset
dog cat mouse elephant77.2 Sample Output
((cat,(mouse,elephant)))dog
((mouse,(cat,elephant)))dog
((elephant,(cat,mouse)))dog77.3 Solution
from itertools import combinations
def generate_combinations(n, items):
"""
Generates all combinations of `n` elements from the list `items`.
"""
if n == 0:
return [[]]
if not items:
return []
head, *tail = items
with_head = [[head] + rest for rest in generate_combinations(n-1, tail)]
without_head = generate_combinations(n, tail)
return with_head + without_head
def memoized_combinations(n, k):
"""
Returns all `k`-combinations of `n` items using memoization.
This prevents redundant computation of the same combination.
"""
memo = memoized_combinations.cache
if n not in memo:
memo[n] = {}
if k not in memo[n]:
memo[n][k] = generate_combinations(k, list(range(n)))
return memo[n][k]
memoized_combinations.cache = {}
def generate_unrooted_binary_trees(species):
"""
Generates all possible unrooted binary trees in Newick format for a given list of species.
"""
if len(species) == 1:
return species
elif len(species) == 2:
return [f"({species[0]},{species[1]})"]
elif len(species) >= 3:
trees = []
for k in range(1, (len(species) // 2) + 1):
seen_combinations = set()
for selected_indices in memoized_combinations(len(species), k):
selected_species = [species[i] for i in selected_indices]
selected_key = ':'.join(sorted(selected_species))
if selected_key in seen_combinations:
continue
seen_combinations.add(selected_key)
remaining_species = [sp for sp in species if sp not in selected_species]
remaining_key = ':'.join(sorted(remaining_species))
if remaining_key in seen_combinations:
continue
seen_combinations.add(remaining_key)
for left_tree in generate_unrooted_binary_trees(remaining_species):
for right_tree in generate_unrooted_binary_trees(selected_species):
trees.append(f"({right_tree},{left_tree})")
return trees
else:
raise Exception("Unexpected number of species")
input_data = "dog cat mouse elephant"
species_list = input_data.split()
root_species = species_list.pop(0)
for tree in generate_unrooted_binary_trees(species_list):
print(f"({tree}){root_species}")77.4 Explanation of the Code
generate_combinationsFunction:- This function is a recursive implementation to generate all possible combinations of
nelements from the given listitems. - It works by considering each element (
head) and recursively generating combinations with (with_head) and without (without_head) that element.
- This function is a recursive implementation to generate all possible combinations of
memoized_combinationsFunction:- This function leverages memoization to store previously computed combinations to avoid redundant calculations.
- It checks if the combination of
nandkhas already been computed and stored in thecache(a dictionary attached to the function). - If not, it computes the combination using
generate_combinationsand stores it for future use.
generate_unrooted_binary_treesFunction:- This function generates all possible unrooted binary trees for the provided list of species in Newick format.
- It uses a recursive approach:
- If there’s only one species, it simply returns it.
- If there are two species, it returns them in a pair.
- If there are three or more species, it splits them into subgroups and recursively generates trees for each subgroup, ensuring that each possible tree structure is considered without duplicates.
78 Genome Assembly Using Reads
A directed cycle is simply a cycle in a directed graph in which the head of one edge is equal to the tail of the next (so that every edge in the cycle is traversed in the same direction).
For a set of DNA strings \(S\) and a positive integer \(k\), let \(Sk\) denote the collection of all possible \(k\)-mers of the strings in \(S\).
Given: A collection \(S\) of (error-free) reads of equal length (not exceeding 50 bp). In this dataset, for some positive integer \(k\), the de Bruijn graph \(Bk\) on \(S_{k+1}∪S^rc_{k+1}\) consists of exactly two directed cycles.
Return: A cyclic superstring of minimal length containing every read or its reverse complement.
78.1 Sample Dataset
AATCT
TGTAA
GATTA
ACAGA78.2 Sample Output
TGTAATC78.3 Solution
from itertools import chain
def reverse_complement(dna):
"""Returns the reverse complement of a given DNA strand."""
translation_table = str.maketrans('ATCG', 'TAGC')
return dna.translate(translation_table)[::-1]
def find_cyclic_superstring(dna_strings):
"""Finds the cyclic superstring from a list of DNA strings and their reverse complements."""
def flatten_list_of_lists(list_of_lists):
"""Flattens one level of nesting in a list of lists."""
return chain.from_iterable(list_of_lists)
num_strings = len(dna_strings)
string_length = len(dna_strings[0]) # Assumes all strings have the same length
for k in range(string_length - 1, 1, -1):
# Create adjacency list of k-mers
adj_list = dict(flatten_list_of_lists([
[(dna[i:i+k], dna[i+1:i+k+1]) for i in range(string_length - k)]
for dna in dna_strings
]))
# Start with an arbitrary k-mer and initialize the superstring
first_kmer = kmer = next(iter(adj_list))
superstring = ''
while True:
if kmer in adj_list:
# Add the last character of the k-mer to the superstring
superstring += kmer[-1]
# Move to the next k-mer in the path
kmer = adj_list.pop(kmer)
# If we have returned to the start, we have completed a cycle
if kmer == first_kmer:
return superstring
else:
# Exit if no continuation of the k-mer path is found
break
# Read and preproces input
sample_input = """
AATCT
TGTAA
GATTA
ACAGA
"""
dna_strings = sample_input.strip().split('\n')
# Add reverse complements of the DNA strings
dna_strings = list(set(dna_strings + [reverse_complement(dna) for dna in dna_strings]))
# Find and print the cyclic superstring
print(find_cyclic_superstring(dna_strings))78.4 Explanation
- Reverse Complement Function:
reverse_complement(dna): Computes the reverse complement of a DNA sequence. It uses a translation table to map each nucleotide to its complement and then reverses the string.
- Finding Cyclic Superstring:
find_cyclic_superstring(dna_strings): Finds a cyclic superstring from the given list of DNA strings. It:- Uses
flatten_list_of_liststo flatten a list of lists into a single list. - Iterates over possible lengths of k-mers from the longest (one les than the length of the DNA strings) to 2.
- Constructs a De Bruijn graph where edges are k-mers, and vertices are k-1-mers.
- Tries to find a cyclic path in the graph that returns to the starting k-mer and constructs the superstring from this path.
- Uses
- Input Processing:
sample_input: Defines a sample input containing multiple DNA strings.dna_strings: Converts the input into a list of DNA strings and adds their reverse complements to handle both possible orientations.
- Finding and Printing the Superstring:
- Calls
find_cyclic_superstringwith the DNA strings (including their reverse complements) to find the cyclic superstring and prints the result.
- Calls
79 Global Alignment with Constant Gap Penalty
In a constant gap penalty, every gap receives some predetermined constant penalty, regardles of its length. Thus, the insertion or deletion of 1000 contiguous symbols is penalized equally to that of a single symbol.
Given: Two protein strings \(s\) and \(t\) in FASTA format (each of length at most 1000 aa).
Return: The maximum alignment score between \(s\) and \(t\). Use:
- The BLOSUM62 scoring matrix.
- Constant gap penalty equal to 5.
79.1 Sample Dataset
>Rosalind_79
PLEASANTLY
>Rosalind_41
MEANLY79.2 Sample Output
1379.3 Solution
BLOSUM62 = {
('W', 'F'): 1, ('L', 'R'): -2, ('S', 'P'): -1, ('V', 'T'): 0,
('Q', 'Q'): 5, ('N', 'A'): -2, ('Z', 'Y'): -2, ('W', 'R'): -3,
('Q', 'A'): -1, ('S', 'D'): 0, ('H', 'H'): 8, ('S', 'H'): -1,
('H', 'D'): -1, ('L', 'N'): -3, ('W', 'A'): -3, ('Y', 'M'): -1,
('G', 'R'): -2, ('Y', 'I'): -1, ('Y', 'E'): -2, ('B', 'Y'): -3,
('Y', 'A'): -2, ('V', 'D'): -3, ('B', 'S'): 0, ('Y', 'Y'): 7,
('G', 'N'): 0, ('E', 'C'): -4, ('Y', 'Q'): -1, ('Z', 'Z'): 4,
('V', 'A'): 0, ('C', 'C'): 9, ('M', 'R'): -1, ('V', 'E'): -2,
('T', 'N'): 0, ('P', 'P'): 7, ('V', 'I'): 3, ('V', 'S'): -2,
('Z', 'P'): -1, ('V', 'M'): 1, ('T', 'F'): -2, ('V', 'Q'): -2,
('K', 'K'): 5, ('P', 'D'): -1, ('I', 'H'): -3, ('I', 'D'): -3,
('T', 'R'): -1, ('P', 'L'): -3, ('K', 'G'): -2, ('M', 'N'): -2,
('P', 'H'): -2, ('F', 'Q'): -3, ('Z', 'G'): -2, ('X', 'L'): -1,
('T', 'M'): -1, ('Z', 'C'): -3, ('X', 'H'): -1, ('D', 'R'): -2,
('B', 'W'): -4, ('X', 'D'): -1, ('Z', 'K'): 1, ('F', 'A'): -2,
('Z', 'W'): -3, ('F', 'E'): -3, ('D', 'N'): 1, ('B', 'K'): 0,
('X', 'X'): -1, ('F', 'I'): 0, ('B', 'G'): -1, ('X', 'T'): 0,
('F', 'M'): 0, ('B', 'C'): -3, ('Z', 'I'): -3, ('Z', 'V'): -2,
('S', 'S'): 4, ('L', 'Q'): -2, ('W', 'E'): -3, ('Q', 'R'): 1,
('N', 'N'): 6, ('W', 'M'): -1, ('Q', 'C'): -3, ('W', 'I'): -3,
('S', 'C'): -1, ('L', 'A'): -1, ('S', 'G'): 0, ('L', 'E'): -3,
('W', 'Q'): -2, ('H', 'G'): -2, ('S', 'K'): 0, ('Q', 'N'): 0,
('N', 'R'): 0, ('H', 'C'): -3, ('Y', 'N'): -2, ('G', 'Q'): -2,
('Y', 'F'): 3, ('C', 'A'): 0, ('V', 'L'): 1, ('G', 'E'): -2,
('G', 'A'): 0, ('K', 'R'): 2, ('E', 'D'): 2, ('Y', 'R'): -2,
('M', 'Q'): 0, ('T', 'I'): -1, ('C', 'D'): -3, ('V', 'F'): -1,
('T', 'A'): 0, ('T', 'P'): -1, ('B', 'P'): -2, ('T', 'E'): -1,
('V', 'N'): -3, ('P', 'G'): -2, ('M', 'A'): -1, ('K', 'H'): -1,
('V', 'R'): -3, ('P', 'C'): -3, ('M', 'E'): -2, ('K', 'L'): -2,
('V', 'V'): 4, ('M', 'I'): 1, ('T', 'Q'): -1, ('I', 'G'): -4,
('P', 'K'): -1, ('M', 'M'): 5, ('K', 'D'): -1, ('I', 'C'): -1,
('Z', 'D'): 1, ('F', 'R'): -3, ('X', 'K'): -1, ('Q', 'D'): 0,
('X', 'G'): -1, ('Z', 'L'): -3, ('X', 'C'): -2, ('Z', 'H'): 0,
('B', 'L'): -4, ('B', 'H'): 0, ('F', 'F'): 6, ('X', 'W'): -2,
('B', 'D'): 4, ('D', 'A'): -2, ('S', 'L'): -2, ('X', 'S'): 0,
('F', 'N'): -3, ('S', 'R'): -1, ('W', 'D'): -4, ('V', 'Y'): -1,
('W', 'L'): -2, ('H', 'R'): 0, ('W', 'H'): -2, ('H', 'N'): 1,
('W', 'T'): -2, ('T', 'T'): 5, ('S', 'F'): -2, ('W', 'P'): -4,
('L', 'D'): -4, ('B', 'I'): -3, ('L', 'H'): -3, ('S', 'N'): 1,
('B', 'T'): -1, ('L', 'L'): 4, ('Y', 'K'): -2, ('E', 'Q'): 2,
('Y', 'G'): -3, ('Z', 'S'): 0, ('Y', 'C'): -2, ('G', 'D'): -1,
('B', 'V'): -3, ('E', 'A'): -1, ('Y', 'W'): 2, ('E', 'E'): 5,
('Y', 'S'): -2, ('C', 'N'): -3, ('V', 'C'): -1, ('T', 'H'): -2,
('P', 'R'): -2, ('V', 'G'): -3, ('T', 'L'): -1, ('V', 'K'): -2,
('K', 'Q'): 1, ('R', 'A'): -1, ('I', 'R'): -3, ('T', 'D'): -1,
('P', 'F'): -4, ('I', 'N'): -3, ('K', 'I'): -3, ('M', 'D'): -3,
('V', 'W'): -3, ('W', 'W'): 11, ('M', 'H'): -2, ('P', 'N'): -2,
('K', 'A'): -1, ('M', 'L'): 2, ('K', 'E'): 1, ('Z', 'E'): 4,
('X', 'N'): -1, ('Z', 'A'): -1, ('Z', 'M'): -1, ('X', 'F'): -1,
('K', 'C'): -3, ('B', 'Q'): 0, ('X', 'B'): -1, ('B', 'M'): -3,
('F', 'C'): -2, ('Z', 'Q'): 3, ('X', 'Z'): -1, ('F', 'G'): -3,
('B', 'E'): 1, ('X', 'V'): -1, ('F', 'K'): -3, ('B', 'A'): -2,
('X', 'R'): -1, ('D', 'D'): 6, ('W', 'G'): -2, ('Z', 'F'): -3,
('S', 'Q'): 0, ('W', 'C'): -2, ('W', 'K'): -3, ('H', 'Q'): 0,
('L', 'C'): -1, ('W', 'N'): -4, ('S', 'A'): 1, ('L', 'G'): -4,
('W', 'S'): -3, ('S', 'E'): 0, ('H', 'E'): 0, ('S', 'I'): -2,
('H', 'A'): -2, ('S', 'M'): -1, ('Y', 'L'): -1, ('Y', 'H'): 2,
('Y', 'D'): -3, ('E', 'R'): 0, ('X', 'P'): -2, ('G', 'G'): 6,
('G', 'C'): -3, ('E', 'N'): 0, ('Y', 'T'): -2, ('Y', 'P'): -3,
('T', 'K'): -1, ('A', 'A'): 4, ('P', 'Q'): -1, ('T', 'C'): -1,
('V', 'H'): -3, ('T', 'G'): -2, ('I', 'Q'): -3, ('Z', 'T'): -1,
('C', 'R'): -3, ('V', 'P'): -2, ('P', 'E'): -1, ('M', 'C'): -1,
('K', 'N'): 0, ('I', 'I'): 4, ('P', 'A'): -1, ('M', 'G'): -3,
('T', 'S'): 1, ('I', 'E'): -3, ('P', 'M'): -2, ('M', 'K'): -1,
('I', 'A'): -1, ('P', 'I'): -3, ('R', 'R'): 5, ('X', 'M'): -1,
('L', 'I'): 2, ('X', 'I'): -1, ('Z', 'B'): 1, ('X', 'E'): -1,
('Z', 'N'): 0, ('X', 'A'): 0, ('B', 'R'): -1, ('B', 'N'): 3,
('F', 'D'): -3, ('X', 'Y'): -1, ('Z', 'R'): 0, ('F', 'H'): -1,
('B', 'F'): -3, ('F', 'L'): 0, ('X', 'Q'): -1, ('B', 'B'): 4
}
def parse_fasta(fasta_string):
"""Parse the input FASTA format string into individual sequences."""
sequences = []
current_sequence = ""
for line in fasta_string.strip().split('\n'):
if line.startswith(">"):
if current_sequence:
sequences.append(current_sequence)
current_sequence = ""
else:
current_sequence += line.strip()
sequences.append(current_sequence) # Append the last sequence
return sequences
def global_alignment_with_gap_penalty(seq1, seq2):
"""Compute the global alignment score between two sequences with gap penalties."""
seq1 = "-" + seq1 # Add a leading gap for alignment
seq2 = "-" + seq2 # Add a leading gap for alignment
# Initialize scoring matrices
lower_matrix = [[0 for _ in range(len(seq2))] for _ in range(len(seq1))]
middle_matrix = [[0 for _ in range(len(seq2))] for _ in range(len(seq1))]
upper_matrix = [[0 for _ in range(len(seq2))] for _ in range(len(seq1))]
# Set gap penalties for the first row and column
for col in range(1, len(seq2)):
lower_matrix[0][col] = -5
middle_matrix[0][col] = -5
upper_matrix[0][col] = -50
for row in range(1, len(seq1)):
lower_matrix[row][0] = -5
middle_matrix[row][0] = -5
upper_matrix[row][0] = -50
# Fill the scoring matrices
for col in range(1, len(seq2)):
for row in range(1, len(seq1)):
lower_matrix[row][col] = max(lower_matrix[row - 1][col], middle_matrix[row - 1][col] - 5)
upper_matrix[row][col] = max(upper_matrix[row][col - 1], middle_matrix[row][col - 1] - 5)
pair = (seq1[row], seq2[col])
reverse_pair = (seq2[col], seq1[row])
score = BLOSUM62.get(pair, BLOSUM62.get(reverse_pair, 0))
middle_matrix[row][col] = max(
lower_matrix[row][col],
middle_matrix[row - 1][col - 1] + score,
upper_matrix[row][col]
)
return middle_matrix[len(seq1) - 1][len(seq2) - 1]
# Sample Input
sample_input = """>Rosalind_79
PLEASANTLY
>Rosalind_41
MEANLY
"""
# Parse input FASTA format data
sequences = parse_fasta(sample_input)
sequence1, sequence2 = sequences[0], sequences[1]
# Compute the global alignment score
alignment_score = global_alignment_with_gap_penalty(sequence1, sequence2)
print(alignment_score)The code computes the global alignment score between two sequences using a scoring matrix (BLOSUM62) and gap penalties. This involves parsing FASTA formatted sequence data, initializing scoring matrices for dynamic programming, and then filling these matrices to calculate the alignment score.
79.4 1. parse_fasta(fasta_string)
Purpose: Parses a FASTA format string into individual sequences.
How it works: - Input: A FASTA format string where sequences are prefixed with a > character. - Output: A list of sequences.
Steps: 1. Initialize an empty list sequences to store parsed sequences. 2. Initialize an empty string current_sequence to build sequences as you read the input. 3. Split the input string into lines and iterate through them: - If a line starts with >, it indicates the start of a new sequence. Append the current sequence to sequences if it’s not empty, then reset current_sequence. - Otherwise, append the line (after stripping whitespace) to current_sequence. 4. Append the last sequence after exiting the loop. 5. Return the list of sequences.
79.5 2. global_alignment_with_gap_penalty(seq1, seq2)
Purpose: Computes the global alignment score between two sequences using a dynamic programming approach with gap penalties.
How it works: - Input: Two sequences, seq1 and seq2. - Output: The alignment score for the sequences.
Steps: 1. Initialize Sequences: - Prepend a gap (-) to both sequences to handle gaps at the start of alignments.
- Initialize Matrices:
lower_matrix,middle_matrix,upper_matrixare 2D lists used to store scores during alignment:lower_matrixtracks scores when aligning with gaps inseq2.upper_matrixtracks scores when aligning with gaps inseq1.middle_matrixstores the scores for the current alignment considering both sequences.
- Set Initial Gap Penalties:
- For the first row (aligning gaps in
seq1with the second sequence), initialize with gap penalties of-5forlower_matrixandmiddle_matrix, and a larger penalty-50forupper_matrix. - For the first column (aligning gaps in
seq2with the first sequence), use similar penalties.
- For the first row (aligning gaps in
- Fill Matrices:
- Use nested loops to fill the matrices:
- Lower Matrix: Computes scores for gaps in
seq2. - Upper Matrix: Computes scores for gaps in
seq1. - Middle Matrix: Computes scores for aligning characters in
seq1andseq2.- Retrieve the score from
BLOSUM62for the pair of characters. - Use the maximum of the scores from
lower_matrix,middle_matrix, andupper_matrixto fill inmiddle_matrix.
- Retrieve the score from
- Lower Matrix: Computes scores for gaps in
- Use nested loops to fill the matrices:
- Retrieve Final Score:
- The final alignment score is in
middle_matrix[len(seq1) - 1][len(seq2) - 1], which represents the best alignment score for the entire length of both sequences.
- The final alignment score is in
80 Inferring Genotype from a Pedigree
A rooted binary tree can be used to model the pedigree of an individual. In this case, rather than time progressing from the root to the leaves, the tree is viewed upside down with time progressing from an individual’s ancestors (at the leaves) to the individual (at the root).
An example of a pedigree for a single factor in which only the genotypes of ancestors are given.
Given: A rooted binary tree \(T\) in Newick format encoding an individual’s pedigree for a Mendelian factor whose alleles are A (dominant) and a (recessive).
Return: Three numbers between 0 and 1, corresponding to the respective probabilities that the individual at the root of \(T\) will exhibit the “AA”, “Aa” and “aa” genotypes.
80.1 Sample Dataset
((((Aa,aa),(Aa,Aa)),((aa,aa),(aa,AA))),Aa);80.2 Sample Output
0.156 0.5 0.34480.3 Solution
# Probabilities from an individual's ancestors (based on Mendelian inheritance)
prob_dict = {
("AA", "AA"): (1.0, 0.0, 0.0),
("AA", "Aa"): (0.5, 0.5, 0.0),
("AA", "aa"): (0.0, 1.0, 0.0),
("Aa", "AA"): (0.5, 0.5, 0.0),
("Aa", "Aa"): (0.25, 0.5, 0.25),
("Aa", "aa"): (0.0, 0.5, 0.5),
("aa", "AA"): (0.0, 1.0, 0.0),
("aa", "Aa"): (0.0, 0.5, 0.5),
("aa", "aa"): (0.0, 0.0, 1.0)
}
def calculate_child_probabilities(parent1, parent2):
"""
Given the genotype probabilities of two parents, calculate the probabilities for the child.
:param parent1: Dictionary containing the probabilities for the first parent (keys: 'AA', 'Aa', 'aa')
:param parent2: Dictionary containing the probabilities for the second parent (keys: 'AA', 'Aa', 'aa')
:return: Dictionary containing the probabilities for the child (keys: 'AA', 'Aa', 'aa')
"""
child_prob = {"AA": 0.0, "Aa": 0.0, "aa": 0.0}
# Combine the genotype probabilities from both parents using the Mendelian inheritance rules
for p1_genotype, p1_prob in parent1.items():
for p2_genotype, p2_prob in parent2.items():
# Look up the probability distribution for the child based on the parent genotypes
child_distribution = prob_dict[(p1_genotype, p2_genotype)]
child_prob["AA"] += p1_prob * p2_prob * child_distribution[0]
child_prob["Aa"] += p1_prob * p2_prob * child_distribution[1]
child_prob["aa"] += p1_prob * p2_prob * child_distribution[2]
return child_prob
# Known genotype probabilities for the leaves (ancestral individuals)
AA = {"AA": 1.0, "Aa": 0.0, "aa": 0.0}
Aa = {"AA": 0.0, "Aa": 1.0, "aa": 0.0}
aa = {"AA": 0.0, "Aa": 0.0, "aa": 1.0}
# Pedigree input in Newick format
pedigree = "((((Aa,aa),(Aa,Aa)),((aa,aa),(aa,AA))),Aa);"
# Modify the Newick string to turn it into a Python expression using the `calculate_child_probabilities` function
pedigree_expression = pedigree.replace(";", "").replace("(", "calculate_child_probabilities(")
# Evaluate the expression to calculate the probabilities for the root individual
root_probabilities = eval(pedigree_expression)
# Print the final probabilities for the root individual
print(f"{root_probabilities['AA']:f} {root_probabilities['Aa']:f} {root_probabilities['aa']:f}")80.4 Explanation
prob_dict:- This dictionary contains the Mendelian inheritance probabilities for a child given the genotypes of both parents. Each tuple of parent genotypes maps to a tuple of probabilities representing the likelihood of the child being
AA,Aa, oraa.
- This dictionary contains the Mendelian inheritance probabilities for a child given the genotypes of both parents. Each tuple of parent genotypes maps to a tuple of probabilities representing the likelihood of the child being
calculate_child_probabilities:- This function calculates the probabilities for a child’s genotype based on the genotype probabilities of their two parents.
- It iterates over all combinations of the parents’ genotypes, looks up the probabilities from
prob_dict, and accumulates the resulting probabilities for the child.
- Leaf Node Probabilities:
- The genotype probabilities for the leaf nodes (the known ancestors) are defined:
AA: 100% chance of beingAAAa: 100% chance of beingAaaa: 100% chance of beingaa
- The genotype probabilities for the leaf nodes (the known ancestors) are defined:
- Pedigree Parsing:
- The input pedigree is provided in Newick format. This format is transformed into a Python expression that can be evaluated using
eval. The key part of this transformation is replacing the(characters with calls to thecalculate_child_probabilitiesfunction. This allows the pedigree to be evaluated as a nested set of function calls, starting from the leaves and working up to the root.
- The input pedigree is provided in Newick format. This format is transformed into a Python expression that can be evaluated using
- Result:
- The program evaluates the pedigree, calculating the probabilities of the root individual being
AA,Aa, oraa. These probabilities are then printed to three decimal places.
- The program evaluates the pedigree, calculating the probabilities of the root individual being
81 Linguistic Complexity of a Genome
Given a length n string s formed over an alphabet \(𝒜\) of size aa, let the “substring count” \(sub(s)\) denote the total number of distinct substrings of \(s\). Furthermore, let the “maximum substring count” \(m(a,n)\) denote the maximum number of distinct substrings that could appear in a string of length \(n\) formed over \(𝒜\).
The linguistic complexity of \(s\) (written \(lc(s)\)) is equal to \(\frac{sub(s)}{m(a,n)}\); in other words, \(lc(s)\) represents the percentage of observed substrings of s to the total number that are theoretically possible. Note that \(0<lc(s)<1\), with smaller values of \(lc(s)\) indicating that s is more repetitive.
As an example, consider the DNA string (\(a=4\)) \(s=ATTTGGATT\). In the following table, we demonstrate that \(lc(s)=\frac{35}{40}=0.875\) by considering the number of observed and possible length \(k\) substrings of \(s\), which are denoted by \(subk(s)\) and \(m(a,k,n)\), respectively. (Observe that \(m(a,n)=/sum^n_{k=1}m(a,k,n)=40\) and \(sub(s)=/sum^n_{k=1}subk(s)=35\))
| kk | subk(s)subk(s) | m(a,k,n)m(a,k,n) |
| 1 | 3 | 4 |
| 2 | 5 | 8 |
| 3 | 6 | 7 |
| 4 | 6 | 6 |
| 5 | 5 | 5 |
| 6 | 4 | 4 |
| 7 | 3 | 3 |
| 8 | 2 | 2 |
| 9 | 1 | 1 |
| Total | 35 | 40 |
Given: A DNA string s of length at most 100 kbp.
Return: The linguistic complexity lc(s)lc(s).
81.1 Sample Dataset
ATTTGGATT81.2 Sample Output
0.87581.3 Solution
import sys
from math import log
clas SuffixTree:
'''Creates a suffix tree for the provided word.'''
def __init__(self, input_word):
'''Initializes the suffix tree.'''
self.nodes = [self.Node(None, 0)]
self.edges = dict()
self.descendants_count = dict()
if isinstance(input_word, str):
self.build_suffix_tree(input_word)
clas Node(object):
'''Suffix tree node class.'''
def __init__(self, parent_node, node_number):
self.parent = parent_node
self.number = node_number
self.children = []
def add_child(self, child_node):
self.children.append(child_node)
def remove_child(self, child_node):
self.children.remove(child_node)
def update_parent(self, new_parent):
self.parent = new_parent
def build_suffix_tree(self, input_word):
'''Build the suffix tree from the input word.'''
if input_word[-1] != '$':
input_word += '$'
self.word = input_word
self.length = len(self.word)
for i in range(self.length):
parent_node, edge_start, overlap_exists = self.find_insert_position(i, self.nodes[0])
if overlap_exists:
old_edge_start, old_edge_end = self.edges[(parent_node.parent.number, parent_node.number)]
# Determine the length of the edge to be inserted
insertion_length = 0
while input_word[edge_start:edge_start + insertion_length] == input_word[old_edge_start:old_edge_start + insertion_length]:
insertion_length += 1
# Create a new node for the insertion
new_node = self.Node(parent_node.parent, len(self.nodes))
new_node.add_child(parent_node)
self.add_edge(parent_node.parent, old_edge_start, old_edge_start + insertion_length - 1, new_node)
# Update the parent node since a new node is inserted above it
del self.edges[(parent_node.parent.number, parent_node.number)]
parent_node.parent.remove_child(parent_node)
parent_node.update_parent(new_node)
self.edges[(parent_node.parent.number, parent_node.number)] = [old_edge_start + insertion_length - 1, old_edge_end]
# Add new child node
self.add_edge(new_node, edge_start + insertion_length - 1, self.length)
else:
# No insertion necessary, just append the new node
self.add_edge(parent_node, edge_start, self.length)
def find_insert_position(self, start_index, parent_node):
'''Finds the appropriate position to insert a suffix into the suffix tree.'''
for child_node in parent_node.children:
edge_start, edge_end = self.edges[(parent_node.number, child_node.number)]
if self.word[start_index:start_index + edge_end - edge_start] == self.word[edge_start:edge_end]:
return self.find_insert_position(start_index + edge_end - edge_start, child_node)
elif self.word[edge_start] == self.word[start_index]:
return child_node, start_index, True
return parent_node, start_index, False
def add_edge(self, parent_node, edge_start, edge_end, child_node=None):
'''Adds a node and the associated edge to the suffix tree.'''
if child_node is None:
child_node = self.Node(parent_node, len(self.nodes))
self.nodes.append(child_node)
parent_node.add_child(child_node)
self.edges[(parent_node.number, child_node.number)] = [edge_start, edge_end]
def get_edge_strings(self):
'''Returns the string representations of the edges.'''
return [self.word[i:j] for i, j in self.edges.values()]
def calculate_descendants(self, base_node):
'''Calculates the total number of descendants of a given node.'''
if base_node not in self.descendants_count:
self.descendants_count[base_node] = len(base_node.children) + sum([self.calculate_descendants(c) for c in base_node.children])
return self.descendants_count[base_node]
def get_node_word(self, end_node):
'''Returns the prefix of the suffix tree word up to a given node.'''
accumulated_word = ''
while end_node.number != 0:
edge_indices = self.edges[(end_node.parent.number, end_node.number)]
accumulated_word = self.word[edge_indices[0]:edge_indices[1]] + accumulated_word
end_node = end_node.parent
return accumulated_word.strip('$')
# Sample input
dna_sequence = "ATTTGGATT"
sequence_length = len(dna_sequence)
# After removing the termination symbol $, if necessary, each edge corresponds to len(edge) substrings
edge_lengths = [edge if edge[1] != sequence_length + 1 else [edge[0], sequence_length] for edge in SuffixTree(dna_sequence).edges.values()]
observed_substrings = float(sum([edge[1] - edge[0] for edge in edge_lengths]))
# The number of possible substrings of length k is min(4^k, n-k-1)
max_possible_substrings = float(sum([sequence_length - k + 1 if k > log(sequence_length + 1) / log(4) else 4 ** k for k in range(1, sequence_length + 1)]))
print(observed_substrings / max_possible_substrings)81.4 Explanation
81.4.1 1. SuffixTree Class
__init__(self, input_word): Initializes the suffix tree with the given word. It sets up initial structures and callsbuild_suffix_treeto construct the tree.NodeClass: Represents nodes in the suffix tree.__init__(self, parent_node, node_number): Initializes a node with a parent and a unique number.add_child(self, child_node): Adds a child node.remove_child(self, child_node): Removes a child node.update_parent(self, new_parent): Updates the parent of the node.
build_suffix_tree(self, input_word): Constructs the suffix tree for the given word. Adds a terminator character$if not already present.find_insert_position(self, start_index, parent_node): Determines where to insert a suffix starting atstart_indexunderparent_node. Handles edge splitting and suffix insertion.add_edge(self, parent_node, edge_start, edge_end, child_node=None): Adds an edge betweenparent_nodeandchild_nodewith start and end indices.get_edge_strings(self): Returns the substrings represented by each edge in the suffix tree.calculate_descendants(self, base_node): Computes the total number of descendants ofbase_node, caching results for efficiency.get_node_word(self, end_node): Constructs the string from the root toend_node.
81.4.2 2. Calculations
observed_substrings: Computes the total length of all distinct substrings from the suffix tree.max_possible_substrings: Calculates the maximum number of distinct substrings possible for a string of lengthnwith an alphabet of size 4.print(observed_substrings / max_possible_substrings): Outputs the linguistic complexity as the ratio of observed distinct substrings to the maximum possible distinct substrings.
82 Local Alignment with Scoring Matrix
A local alignment of two strings \(s\) and \(t\) is an alignment of substrings rr and uu of \(s\) and \(t\), respectively. Let \(opt(r,u)\) denote the score of an optimal alignment of \(r\) and \(u\) with respect to some predetermined alignment score.
Given: Two protein strings \(s\) and \(t\) in FASTA format (each having length at most 1000 aa).
Return: A maximum alignment score along with substrings \(r\) and \(u\) of \(s\) and \(t\), respectively, which produce this maximum alignment score (multiple solutions may exist, in which case you may output any one).
Use: - The PAM250 scoring matrix. - Linear gap penalty equal to 5.
82.1 Sample Dataset
>Rosalind_80
MEANLYPRTEINSTRING
>Rosalind_21
PLEASANTLYEINSTEIN82.2 Sample Output
23
LYPRTEINSTRIN
LYEINSTEIN82.3 Solution
import numpy as np
clas PAM250:
"""The PAM250 scoring matrix class."""
def __init__(self):
"""Initialize the scoring matrix."""
# Manually define the PAM250 scoring matrix.
self.scoring_matrix = {
('A', 'A'): 2, ('A', 'C'): -2, ('A', 'D'): 0, ('A', 'E'): 0, ('A', 'F'): -3, ('A', 'G'): 1, ('A', 'H'): -1,
('A', 'I'): -1, ('A', 'K'): -1, ('A', 'L'): -2, ('A', 'M'): -1, ('A', 'N'): 0, ('A', 'P'): 1, ('A', 'Q'): 0,
('A', 'R'): -2, ('A', 'S'): 1, ('A', 'T'): 1, ('A', 'V'): 0, ('A', 'W'): -6, ('A', 'Y'): -3,
('C', 'A'): -2, ('C', 'C'): 12, ('C', 'D'): -5, ('C', 'E'): -5, ('C', 'F'): -4, ('C', 'G'): -3, ('C', 'H'): -3,
('C', 'I'): -2, ('C', 'K'): -5, ('C', 'L'): -6, ('C', 'M'): -5, ('C', 'N'): -4, ('C', 'P'): -3, ('C', 'Q'): -5,
('C', 'R'): -4, ('C', 'S'): 0, ('C', 'T'): -2, ('C', 'V'): -2, ('C', 'W'): -8, ('C', 'Y'): 0,
('D', 'A'): 0, ('D', 'C'): -5, ('D', 'D'): 4, ('D', 'E'): 3, ('D', 'F'): -6, ('D', 'G'): 1, ('D', 'H'): 1,
('D', 'I'): -2, ('D', 'K'): 0, ('D', 'L'): -4, ('D', 'M'): -3, ('D', 'N'): 2, ('D', 'P'): -1, ('D', 'Q'): 2,
('D', 'R'): -1, ('D', 'S'): 0, ('D', 'T'): 0, ('D', 'V'): -2, ('D', 'W'): -7, ('D', 'Y'): -4,
('E', 'A'): 0, ('E', 'C'): -5, ('E', 'D'): 3, ('E', 'E'): 4, ('E', 'F'): -5, ('E', 'G'): 0, ('E', 'H'): 1,
('E', 'I'): -2, ('E', 'K'): 0, ('E', 'L'): -3, ('E', 'M'): -2, ('E', 'N'): 1, ('E', 'P'): -1, ('E', 'Q'): 2,
('E', 'R'): -1, ('E', 'S'): 0, ('E', 'T'): 0, ('E', 'V'): -2, ('E', 'W'): -7, ('E', 'Y'): -4,
('F', 'A'): -3, ('F', 'C'): -4, ('F', 'D'): -6, ('F', 'E'): -5, ('F', 'F'): 9, ('F', 'G'): -5, ('F', 'H'): -2,
('F', 'I'): 1, ('F', 'K'): -5, ('F', 'L'): 2, ('F', 'M'): 0, ('F', 'N'): -3, ('F', 'P'): -5, ('F', 'Q'): -5,
('F', 'R'): -4, ('F', 'S'): -3, ('F', 'T'): -3, ('F', 'V'): -1, ('F', 'W'): 0, ('F', 'Y'): 7,
('G', 'A'): 1, ('G', 'C'): -3, ('G', 'D'): 1, ('G', 'E'): 0, ('G', 'F'): -5, ('G', 'G'): 5, ('G', 'H'): -2,
('G', 'I'): -3, ('G', 'K'): -2, ('G', 'L'): -4, ('G', 'M'): -3, ('G', 'N'): 0, ('G', 'P'): 0, ('G', 'Q'): -1,
('G', 'R'): -3, ('G', 'S'): 1, ('G', 'T'): 0, ('G', 'V'): -1, ('G', 'W'): -7, ('G', 'Y'): -5,
('H', 'A'): -1, ('H', 'C'): -3, ('H', 'D'): 1, ('H', 'E'): 1, ('H', 'F'): -2, ('H', 'G'): -2, ('H', 'H'): 6,
('H', 'I'): -2, ('H', 'K'): 0, ('H', 'L'): -2, ('H', 'M'): -2, ('H', 'N'): 2, ('H', 'P'): 0, ('H', 'Q'): 3,
('H', 'R'): 2, ('H', 'S'): -1, ('H', 'T'): -1, ('H', 'V'): -2, ('H', 'W'): -3, ('H', 'Y'): 0,
('I', 'A'): -1, ('I', 'C'): -2, ('I', 'D'): -2, ('I', 'E'): -2, ('I', 'F'): 1, ('I', 'G'): -3, ('I', 'H'): -2,
('I', 'I'): 5, ('I', 'K'): -2, ('I', 'L'): 2, ('I', 'M'): 2, ('I', 'N'): -2, ('I', 'P'): -2, ('I', 'Q'): -2,
('I', 'R'): -2, ('I', 'S'): -1, ('I', 'T'): 0, ('I', 'V'): 4, ('I', 'W'): -5, ('I', 'Y'): -1,
('K', 'A'): -1, ('K', 'C'): -5, ('K', 'D'): 0, ('K', 'E'): 0, ('K', 'F'): -5, ('K', 'G'): -2, ('K', 'H'): 0,
('K', 'I'): -2, ('K', 'K'): 5, ('K', 'L'): -3, ('K', 'M'): 0, ('K', 'N'): 1, ('K', 'P'): -1, ('K', 'Q'): 1,
('K', 'R'): 3, ('K', 'S'): 0, ('K', 'T'): 0, ('K', 'V'): -2, ('K', 'W'): -3, ('K', 'Y'): -4,
('L', 'A'): -2, ('L', 'C'): -6, ('L', 'D'): -4, ('L', 'E'): -3, ('L', 'F'): 2, ('L', 'G'): -4, ('L', 'H'): -2,
('L', 'I'): 2, ('L', 'K'): -3, ('L', 'L'): 6, ('L', 'M'): 4, ('L', 'N'): -3, ('L', 'P'): -3, ('L', 'Q'): -2,
('L', 'R'): -3, ('L', 'S'): -3, ('L', 'T'): -2, ('L', 'V'): 2, ('L', 'W'): -2, ('L', 'Y'): -1,
('M', 'A'): -1, ('M', 'C'): -5, ('M', 'D'): -3, ('M', 'E'): -2, ('M', 'F'): 0, ('M', 'G'): -3, ('M', 'H'): -2,
('M', 'I'): 2, ('M', 'K'): 0, ('M', 'L'): 4, ('M', 'M'): 6, ('M', 'N'): -2, ('M', 'P'): -2, ('M', 'Q'): -1,
('M', 'R'): 0, ('M', 'S'): -2, ('M', 'T'): -1, ('M', 'V'): 2, ('M', 'W'): -4, ('M', 'Y'): -2,
('N', 'A'): 0, ('N', 'C'): -4, ('N', 'D'): 2, ('N', 'E'): 1, ('N', 'F'): -3, ('N', 'G'): 0, ('N', 'H'): 2,
('N', 'I'): -2, ('N', 'K'): 1, ('N', 'L'): -3, ('N', 'M'): -2, ('N', 'N'): 2, ('N', 'P'): 0, ('N', 'Q'): 1,
('N', 'R'): 0, ('N', 'S'): 1, ('N', 'T'): 0, ('N', 'V'): -2, ('N', 'W'): -4, ('N', 'Y'): -2,
('P', 'A'): 1, ('P', 'C'): -3, ('P', 'D'): -1, ('P', 'E'): -1, ('P', 'F'): -5, ('P', 'G'): 0, ('P', 'H'): 0,
('P', 'I'): -2, ('P', 'K'): -1, ('P', 'L'): -3, ('P', 'M'): -2, ('P', 'N'): 0, ('P', 'P'): 6, ('P', 'Q'): 0,
('P', 'R'): 0, ('P', 'S'): 1, ('P', 'T'): 0, ('P', 'V'): -1, ('P', 'W'): -6, ('P', 'Y'): -5,
('Q', 'A'): 0, ('Q', 'C'): -5, ('Q', 'D'): 2, ('Q', 'E'): 2, ('Q', 'F'): -5, ('Q', 'G'): -1, ('Q', 'H'): 3,
('Q', 'I'): -2, ('Q', 'K'): 1, ('Q', 'L'): -2, ('Q', 'M'): -1, ('Q', 'N'): 1, ('Q', 'P'): 0, ('Q', 'Q'): 4,
('Q', 'R'): 1, ('Q', 'S'): -1, ('Q', 'T'): -1, ('Q', 'V'): -2, ('Q', 'W'): -5, ('Q', 'Y'): -4,
('R', 'A'): -2, ('R', 'C'): -4, ('R', 'D'): -1, ('R', 'E'): -1, ('R', 'F'): -4, ('R', 'G'): -3, ('R', 'H'): 2,
('R', 'I'): -2, ('R', 'K'): 3, ('R', 'L'): -3, ('R', 'M'): 0, ('R', 'N'): 0, ('R', 'P'): 0, ('R', 'Q'): 1,
('R', 'R'): 6, ('R', 'S'): 0, ('R', 'T'): -1, ('R', 'V'): -2, ('R', 'W'): 2, ('R', 'Y'): -4,
('S', 'A'): 1, ('S', 'C'): 0, ('S', 'D'): 0, ('S', 'E'): 0, ('S', 'F'): -3, ('S', 'G'): 1, ('S', 'H'): -1,
('S', 'I'): -1, ('S', 'K'): 0, ('S', 'L'): -3, ('S', 'M'): -2, ('S', 'N'): 1, ('S', 'P'): 1, ('S', 'Q'): -1,
('S', 'R'): 0, ('S', 'S'): 2, ('S', 'T'): 1, ('S', 'V'): -1, ('S', 'W'): -2, ('S', 'Y'): -3,
('T', 'A'): 1, ('T', 'C'): -2, ('T', 'D'): 0, ('T', 'E'): 0, ('T', 'F'): -3, ('T', 'G'): 0, ('T', 'H'): -1,
('T', 'I'): 0, ('T', 'K'): 0, ('T', 'L'): -2, ('T', 'M'): -1, ('T', 'N'): 0, ('T', 'P'): 0, ('T', 'Q'): -1,
('T', 'R'): -1, ('T', 'S'): 1, ('T', 'T'): 3, ('T', 'V'): 0, ('T', 'W'): -5, ('T', 'Y'): -3,
('V', 'A'): 0, ('V', 'C'): -2, ('V', 'D'): -2, ('V', 'E'): -2, ('V', 'F'): -1, ('V', 'G'): -1, ('V', 'H'): -2,
('V', 'I'): 4, ('V', 'K'): -2, ('V', 'L'): 2, ('V', 'M'): 2, ('V', 'N'): -2, ('V', 'P'): -1, ('V', 'Q'): -2,
('V', 'R'): -2, ('V', 'S'): -1, ('V', 'T'): 0, ('V', 'V'): 4, ('V', 'W'): -6, ('V', 'Y'): -2,
('W', 'A'): -6, ('W', 'C'): -8, ('W', 'D'): -7, ('W', 'E'): -7, ('W', 'F'): 0, ('W', 'G'): -7, ('W', 'H'): -3,
('W', 'I'): -5, ('W', 'K'): -3, ('W', 'L'): -2, ('W', 'M'): -4, ('W', 'N'): -4, ('W', 'P'): -6, ('W', 'Q'): -5,
('W', 'R'): 2, ('W', 'S'): -2, ('W', 'T'): -5, ('W', 'V'): -6, ('W', 'W'): 17, ('W', 'Y'): 0,
('Y', 'A'): -3, ('Y', 'C'): 0, ('Y', 'D'): -4, ('Y', 'E'): -4, ('Y', 'F'): 7, ('Y', 'G'): -5, ('Y', 'H'): 0,
('Y', 'I'): -1, ('Y', 'K'): -4, ('Y', 'L'): -1, ('Y', 'M'): -2, ('Y', 'N'): -2, ('Y', 'P'): -5, ('Y', 'Q'): -4,
('Y', 'R'): -4, ('Y', 'S'): -3, ('Y', 'T'): -3, ('Y', 'V'): -2, ('Y', 'W'): 0, ('Y', 'Y'): 10
}
def __getitem__(self, pair):
"""Return the score for a given pair of amino acids."""
return self.scoring_matrix[pair]
def local_alignment(v, w, scoring_matrix, sigma):
"""Returns the score and local alignment with the given scoring matrix and indel penalty sigma for strings v, w."""
# Initialize the matrices S (score) and backtrack.
S = np.zeros((len(v) + 1, len(w) + 1), dtype=int)
backtrack = np.zeros((len(v) + 1, len(w) + 1), dtype=int)
# Fill in the score matrix S and the backtrack matrix.
for i in range(1, len(v) + 1):
for j in range(1, len(w) + 1):
match_score = scoring_matrix[v[i - 1], w[j - 1]]
scores = [
S[i - 1][j] - sigma, # Insertion
S[i][j - 1] - sigma, # Deletion
S[i - 1][j - 1] + match_score, # Match/Mismatch
0 # Local alignment can start anywhere, so 0 is a valid score.
]
S[i][j] = max(scores) # Maximum score for cell (i, j)
backtrack[i][j] = scores.index(S[i][j]) # Record which move was taken.
# Find the position of the highest scoring cell in the matrix.
max_pos = np.unravel_index(np.argmax(S), S.shape)
max_score = str(S[max_pos])
# Start backtracking from the position of the highest score.
i, j = max_pos
v_aligned, w_aligned = [], []
# Reconstruct the alignment.
while S[i][j] != 0:
if backtrack[i][j] == 0: # Insertion
i -= 1
v_aligned.append(v[i])
w_aligned.append('-')
elif backtrack[i][j] == 1: # Deletion
j -= 1
v_aligned.append('-')
w_aligned.append(w[j])
else: # Match or mismatch
i -= 1
j -= 1
v_aligned.append(v[i])
w_aligned.append(w[j])
# Reverse the aligned sequences since they were constructed backwards.
v_aligned = ''.join(v_aligned[::-1])
w_aligned = ''.join(w_aligned[::-1])
return max_score, v_aligned, w_aligned
# Sample Input
sample_input = """>Rosalind_80
MEANLYPRTEINSTRING
>Rosalind_21
PLEASANTLYEINSTEIN
"""
# Parse input FASTA format data
def parse_fasta(data):
sequences = []
parts = data.strip().split('>')
for part in parts:
if part:
lines = part.split('\n')
sequence = ''.join(lines[1:])
sequences.append(sequence)
return sequences
sequences = parse_fasta(sample_input)
seq1, seq2 = sequences[0], sequences[1]
# Get the local alignment with sigma = 5.
alignment = local_alignment(seq1, seq2, PAM250(), 5)
# Print the alignment.
print('\n'.join(alignment))82.4 Explanation
- PAM250 Class:
- The
PAM250clas is defined to encapsulate the scoring matrix. The matrix is stored in a dictionary where the keys are tuples of amino acid pairs, and the values are their corresponding scores. - The
__getitem__method allows easy acces to the matrix using tuple indexing, e.g.,scoring_matrix['A', 'A'].
- The
- local_alignment Function:
- The
local_alignmentfunction computes the local alignment using dynamic programming. Sis the score matrix that keeps track of the best scores for subsequences ofvandw.backtrackkeeps track of the choices made to achieve the score in each cell (insertion, deletion, match/mismatch, or starting a new local alignment).- The function iterates over the strings
vandw, filling the matrices according to the local alignment rules. - The backtracking step reconstructs the aligned sequences from the highest scoring position until a score of zero is encountered, indicating the start of the local alignment.
- The
- Backtracking and Alignment:
- The backtracking loop constructs the aligned sequences by following the recorded moves in the
backtrackmatrix. - The sequences are built in reverse and then reversed at the end to give the correct alignment.
- The backtracking loop constructs the aligned sequences by following the recorded moves in the
- parse_fasta:
- The
parse_fastafunction reads the input FASTA format data and extracts the sequences. It returns a list of sequences to be used in the alignment.
- The
- Execution:
- The code parses the sample input, computes the local alignment, and prints the aligned sequences and the alignment score.
83 Maximizing the Gap Symbols of an Optimal Alignment
For the computation of an alignment score generalizing the edit alignment score, let \(m\) denote the score assigned to matched symbols, \(d\) denote the score assigned to mismatched non-gap symbols, and \(g\) denote the score assigned a symbol matched to a gap symbol ‘-’ (i.e., \(g\) is a linear gap penalty).
Given: Two DNA strings \(s\) and \(t\) in FASTA format (each of length at most 5000 bp).
Return: The maximum number of gap symbols that can appear in any maximum score alignment of \(s\) and \(t\) with score parameters satisfying \(m>0\), \(d<0\), and \(g<0\).
83.1 Sample Dataset
>Rosalind_92
AACGTA
>Rosalind_47
ACACCTA83.2 Sample Output
383.3 Solution
def maximum_gap_symbols(v, w):
"""
Returns the maximum number of gap symbols in an optimal alignment of v and w.
The maximum number of gaps is derived from the lengths of the input strings and the longest common subsequence (LCS) between them.
"""
# Initialize a matrix to store the lengths of the longest common subsequence
len_v, len_w = len(v), len(w)
lcs_matrix = [[0] * (len_w + 1) for _ in range(len_v + 1)]
# Fill the matrix based on LCS dynamic programming
for i in range(1, len_v + 1):
for j in range(1, len_w + 1):
if v[i - 1] == w[j - 1]:
lcs_matrix[i][j] = lcs_matrix[i - 1][j - 1] + 1
else:
lcs_matrix[i][j] = max(lcs_matrix[i][j - 1], lcs_matrix[i - 1][j])
# Calculate the maximum number of gap symbols
lcs_length = lcs_matrix[len_v][len_w]
max_gaps = len_v + len_w - 2 * lcs_length
return max_gaps
def parse_fasta(data):
"""
Parses input FASTA format data and returns a list of sequences.
"""
sequences = []
entries = data.strip().split('>')
for entry in entries:
if entry:
lines = entry.splitlines()
sequence = ''.join(lines[1:])
sequences.append(sequence)
return sequences
# Sample input in FASTA format
sample_input = """
>Rosalind_92
AACGTA
>Rosalind_47
ACACCTA
"""
# Parse the sequences from the sample input
sequences = parse_fasta(sample_input)
v, w = sequences[0], sequences[1]
# Get the maximum number of gap symbols
max_gaps = maximum_gap_symbols(v, w)
# Print the result
print(max_gaps)83.4 Explanation
maximum_gap_symbols(v, w) function calculates the maximum number of gap symbols that can appear in any optimal alignment of two DNA strings v and w. It does so by using the Longest Common Subsequence (LCS) method.
Initialization:
len_v, len_w = len(v), len(w) lcs_matrix = [[0] * (len_w + 1) for _ in range(len_v + 1)]Here, the lengths of the input strings
vandware stored inlen_vandlen_w. Then, a 2D matrixlcs_matrixis initialized with all elements set to 0. The matrix has dimensions(len_v + 1) x (len_w + 1).Filling the LCS Matrix:
for i in range(1, len_v + 1): for j in range(1, len_w + 1): if v[i - 1] == w[j - 1]: lcs_matrix[i][j] = lcs_matrix[i - 1][j - 1] + 1 else: lcs_matrix[i][j] = max(lcs_matrix[i][j - 1], lcs_matrix[i - 1][j])This loop fills the
lcs_matrixbased on the LCS dynamic programming approach:- If the characters
v[i-1]andw[j-1]are equal, the value atlcs_matrix[i][j]is updated tolcs_matrix[i-1][j-1] + 1. - Otherwise, it takes the maximum of the values to the left (
lcs_matrix[i][j-1]) and above (lcs_matrix[i-1][j]).
- If the characters
Calculating Maximum Number of Gaps:
lcs_length = lcs_matrix[len_v][len_w] max_gaps = len_v + len_w - 2 * lcs_lengthAfter filling the matrix, the length of the LCS is stored in
lcs_length. The maximum number of gaps is then calculated using the formulalen(v) + len(w) - 2 * lcs_length.
84 Identifying Maximal Repeats
A maximal repeat of a string \(s\) is a repeated substring \(t\) of \(s\) having two occurrences \(t_1\) and \(t_2\) such that \(t_1\) and \(t_2\) cannot be extended by one symbol in either direction in \(s\) and still agree.
For example, “AG” is a maximal repeat in “TAGTTAGCGAGA” because even though the first two occurrences of “AG” can be extended left into “TAG”, the first and third occurrences differ on both sides of the repeat; thus, we conclude that “AG” is a maximal repeat. Note that “TAG” is also a maximal repeat of “TAGTTAGCGAGA”, since its only two occurrences do not still match if we extend them in either direction.
Given: A DNA string \(s\) of length at most 1 kbp.
Return: A list containing all maximal repeats of \(s\) having length at least 20.
84.1 Sample Dataset
TAGAGATAGAATGGGTCCAGAGTTTTGTAATTTCCATGGGTCCAGAGTTTTGTAATTTATTATATAGAGATAGAATGGGTCCAGAGTTTTGTAATTTCCATGGGTCCAGAGTTTTGTAATTTAT84.2 Sample Output
ATGGGTCCAGAGTTTTGTAATTT
TAGAGATAGAATGGGTCCAGAGTTTTGTAATTTCCATGGGTCCAGAGTTTTGTAATTTAT84.3 Solution
import functools
clas SuffixTree(object):
'''Creates a suffix tree for the provided word.'''
def __init__(self, word):
'''Initializes the suffix tree.'''
self.nodes = [self.Node(None, 0)]
self.edges = dict()
self.descendants_dict = dict()
if type(word) == str:
self.add_word(word)
clas Node(object):
'''Suffix tree node class.'''
def __init__(self, parent, number):
self.parent = parent
self.number = number
self.children = []
def add_child(self, child):
self.children.append(child)
def remove_child(self, child):
self.children.remove(child)
def update_parent(self, parent):
self.parent = parent
def add_word(self, word):
'''Add a word to the suffix tree.'''
# Check to make sure word ends in '$'.
if word[-1] != '$':
word += '$'
self.word = word
self.n = len(self.word)
for i in range(self.n):
parent_node, edge_start, overlap = self.insert_position(i, self.nodes[0])
if overlap:
p_edge_start, p_edge_end = self.edges[(parent_node.parent.number, parent_node.number)]
# Get the edge to insert
insert_len = 0
while word[edge_start:edge_start + insert_len] == word[p_edge_start:p_edge_start + insert_len]:
insert_len += 1
# Create a new node for insertion
new_node = self.Node(parent_node.parent, len(self.nodes))
new_node.add_child(parent_node)
self.add_node(parent_node.parent, p_edge_start, p_edge_start + insert_len - 1, new_node)
# Update the parent node since a new node is inserted above it
del self.edges[(parent_node.parent.number, parent_node.number)]
parent_node.parent.remove_child(parent_node)
parent_node.update_parent(new_node)
self.edges[(parent_node.parent.number, parent_node.number)] = [p_edge_start + insert_len - 1, p_edge_end]
# Add new child node
self.add_node(new_node, edge_start + insert_len - 1, self.n)
else:
# No insertion necessary, just append the new node.
self.add_node(parent_node, edge_start, self.n)
def insert_position(self, start_index, parent_node):
'''Determine the location and method to insert a suffix into the suffix tree.'''
for child_node in parent_node.children:
edge_start, edge_end = self.edges[(parent_node.number, child_node.number)]
if self.word[start_index:start_index + edge_end - edge_start] == self.word[edge_start:edge_end]:
return self.insert_position(start_index + edge_end - edge_start, child_node)
elif self.word[edge_start] == self.word[start_index]:
return child_node, start_index, True
return parent_node, start_index, False
def add_node(self, parent_node, edge_start, edge_end, child_node=None):
'''Adds a node and the associated edge to the suffix tree.'''
# Create child node, if necessary
if child_node is None:
child_node = self.Node(parent_node, len(self.nodes))
# Add node to node list
self.nodes.append(child_node)
# Add child to parent
parent_node.add_child(child_node)
# Add edge to edge dict
self.edges[(parent_node.number, child_node.number)] = [
edge_start, edge_end]
def print_edges(self):
'''Returns the string representations of the edges.'''
return [self.word[i:j] for i, j in self.edges.values()]
def total_descendants(self, base_node):
'''Returns the total number of descendants of a given node.'''
if base_node not in self.descendants_dict:
self.descendants_dict[base_node] = len(base_node.children) + sum([self.total_descendants(c) for c in base_node.children])
return self.descendants_dict[base_node]
def node_word(self, end_node):
'''Returns the prefix of the suffix tree word up to a given node.'''
current_word = ''
while end_node.number != 0:
temp_indices = self.edges[(end_node.parent.number, end_node.number)]
current_word = self.word[temp_indices[0]:temp_indices[1]] + current_word
end_node = end_node.parent
return current_word.strip('$')
clas Trie(object):
'''Constructs a trie.'''
def __init__(self, word=None):
self.nodes = [[self.Node('', 1)]]
self.edges = []
if word is not None:
self.add_word(word)
clas Node(object):
'''Trie node class.'''
def __init__(self, prefix, number):
self.prefix = prefix
self.number = number
self.depth = len(prefix)
clas Edge(object):
'''Trie edge class.'''
def __init__(self, letter, par_node, chi_node):
self.letter = letter
self.parent_node = par_node
self.child_node = chi_node
def get_info(self):
'''Return the edge information compactly.'''
return ' '.join(map(str, [self.parent_node, self.child_node, self.letter]))
def add_word(self, word):
'''Adds a word to the trie.'''
if type(word) == list:
for w in word:
self.add_word(w)
else:
parent = self.find_parent(word)
for i in range(len(parent.prefix), len(word)):
new_node = self.Node(word[:i + 1], self.node_count() + 1)
self.edges.append(self.Edge(word[i], parent.number, self.node_count() + 1))
self.insert_node(new_node)
parent = new_node
def insert_node(self, node):
'''Determine the location to insert the current node.'''
if node.depth > self.depth():
self.nodes.append([node])
else:
self.nodes[node.depth].append(node)
def depth(self):
'''Returns the depth of the trie.'''
return len(self.nodes) - 1
def node_count(self):
'''Returns the total number of nodes.'''
count = 0
for trie_depth in self.nodes:
count += len(trie_depth)
return count
def find_parent(self, word):
'''Return the parent node of the word to be inserted.'''
for i in range(min(len(word), self.depth()), 0, -1):
for node in self.nodes[i]:
if word[:i] == node.prefix:
return node
return self.nodes[0][0]
# Read the input data.
sample_input = """
TAGAGATAGAATGGGTCCAGAGTTTTGTAATTTCCATGGGTCCAGAGTTTTGTAATTTATTATATAGAGATAGAATGGGTCCAGAGTTTTGTAATTTCCATGGGTCCAGAGTTTTGTAATTTAT
"""
dna = sample_input.strip()
# Create the Suffix Tree.
suff = SuffixTree(dna)
# Store all multiple repeats of length at least 20 in a dictionary keyed on number of appearances.
repeat_dict = {}
for node in suff.nodes[1:]:
if suff.total_descendants(node) >= 2 and len(suff.node_word(node)) >= 20:
if suff.total_descendants(node) not in repeat_dict:
repeat_dict[suff.total_descendants(node)] = [suff.node_word(node)]
else:
repeat_dict[suff.total_descendants(node)].append(suff.node_word(node))
# Filter out non-maximal repeats.
repeats = []
for values in repeat_dict.values():
if len(values) == 1:
repeats += values
else:
repeats += filter(lambda v: all(v not in word for word in values if word != v), values)
# Print and save the answer.
print('\n'.join(repeats))84.4 Detailed Breakdown of the Code
suff = SuffixTree(dna):- Constructs a suffix tree for the DNA sequence.
Building
repeat_dict:- Collects all repeats that appear at least twice and are at least 20 characters long.
- Uses the
total_descendantsmethod to count occurrences.
Filtering Non-Maximal Repeats:
- Ensures that each repeat is maximal by checking if it is not a substring of any other repeat in the same list.
Printing Results:
- Prints the filtered list of maximal repeats.
85 Multiple Alignment
A multiple alignment of a collection of three or more strings is formed by adding gap symbols to the strings to produce a collection of augmented strings all having the same length.
A multiple alignment score is obtained by taking the sum of an alignment score over all possible pairs of augmented strings. The only difference in scoring the alignment of two strings is that two gap symbols may be aligned for a given pair (requiring us to specify a score for matched gap symbols).
Given: A collection of four DNA strings of length at most 10 bp in FASTA format.
Return: A multiple alignment of the strings having maximum score, where we score matched symbols 0 (including matched gap symbols) and all mismatched symbols -1 (thus incorporating a linear gap penalty of 1).
85.1 Sample Dataset
>Rosalind_7
ATATCCG
>Rosalind_35
TCCG
>Rosalind_23
ATGTACTG
>Rosalind_44
ATGTCTG85.2 Sample Output
-18
ATAT-CCG
-T---CCG
ATGTACTG
ATGT-CTG85.3 Solution
import numpy as np
def score(chars, match=0, mismatch=-1):
"""
Calculate the alignment score for a list of characters.
:param chars: List of characters.
:param match: Score for matching characters.
:param mismatch: Score for mismatching characters.
:return: Total alignment score.
"""
return sum(match if chars[i] == chars[j] else mismatch for i in range(len(chars)) for j in range(i + 1, len(chars)))
def generate_indices(dimensions):
"""
Generate all possible indices for the given dimensions.
:param dimensions: List of dimensions for each sequence.
:return: Generator yielding tuples of indices.
"""
total_combinations = np.prod(dimensions)
indices = [0] * len(dimensions)
for _ in range(total_combinations):
yield tuple(indices)
for j in reversed(range(len(dimensions))):
indices[j] += 1
if indices[j] < dimensions[j]:
break
indices[j] = 0
def generate_moves(num_sequences, options=[0, -1]):
"""
Generate all valid move combinations.
:param num_sequences: Number of sequences.
:param options: Possible move options (0 for match, -1 for gap).
:return: List of valid move combinations.
"""
def recursive_moves(m):
if m == 1:
return [[o] for o in options]
return [[o] + rest for o in options for rest in recursive_moves(m - 1)]
return [move for move in recursive_moves(num_sequences) if any(x != 0 for x in move)]
def add_tuples(u, v):
"""
Add two tuples element-wise.
:param u: First tuple.
:param v: Second tuple.
:return: Element-wise sum of the tuples.
"""
return tuple(a + b for a, b in zip(u, v))
def build_scoring_matrix(Strings, score_function=score):
"""
Build the scoring matrix for the alignment.
:param Strings: List of sequences to align.
:param score_function: Function to calculate alignment score.
:return: Scoring matrix, path dictionary, and move list.
"""
def calculate_scores(index):
def get_score(move):
previous = add_tuples(index, move)
if any(p < 0 for p in previous):
return None
scorable = [Strings[j][previous[j]] if move[j] < 0 else '-' for j in range(len(move))]
return scoring_matrix[previous] + score_function(scorable)
raw_scores = [(get_score(move), move) for move in available_moves]
return [(score, move) for score, move in raw_scores if score is not None]
dimensions = [len(S) + 1 for S in Strings]
scoring_matrix = np.zeros(dimensions, dtype=int)
path = {}
available_moves = generate_moves(len(Strings))
for index_set in generate_indices(dimensions):
scores_moves = calculate_scores(index_set)
if scores_moves:
scores, moves = zip(*scores_moves)
best_index = np.argmax(scores)
scoring_matrix[index_set] = scores[best_index]
path[index_set] = moves[best_index]
return scoring_matrix, path, available_moves
def backtrack_alignment(scoring_matrix, path, Strings):
"""
Perform backtracking to retrieve the optimal alignment.
:param scoring_matrix: Scoring matrix.
:param path: Path dictionary for moves.
:param Strings: List of sequences to align.
:return: Alignment score and aligned sequences.
"""
def reverse_string(s):
return ''.join(reversed(s))
position = tuple(len(S) for S in Strings)
alignment_score = scoring_matrix[position]
alignments = [[] for _ in Strings]
while any(p != 0 for p in position):
move = path[position]
for i, m in enumerate(move):
if m == 0:
alignments[i].append('-')
else:
alignments[i].append(Strings[i][position[i] - 1])
position = add_tuples(position, move)
return alignment_score, [reverse_string(s) for s in alignments]
def FindHighestScoringMultipleSequenceAlignment(Strings, score_function=score):
"""
Find the highest scoring multiple sequence alignment.
:param Strings: List of sequences to align.
:param score_function: Function to calculate alignment score.
:return: Alignment score and aligned sequences.
"""
scoring_matrix, path, _ = build_scoring_matrix(Strings, score_function)
return backtrack_alignment(scoring_matrix, path, Strings)
def parse_fasta(data):
"""
Parse FASTA format data into a list of sequences.
:param data: FASTA format input data.
:return: List of sequences.
"""
sequences = []
entries = data.strip().split('>')
for entry in entries:
if entry:
lines = entry.splitlines()
sequence = ''.join(lines[1:])
sequences.append(sequence)
return sequences
# Sample input in FASTA format
sample_input = """
>Rosalind_7
ATATCCG
>Rosalind_35
TCCG
>Rosalind_23
ATGTACTG
>Rosalind_44
ATGTCTG
"""
# Parse the sequences from the sample input
words = parse_fasta(sample_input)
# Get the alignment.
score, alignment = FindHighestScoringMultipleSequenceAlignment(words)
# Print the alignment score and sequences.
print(score)
for line in alignment:
print(line)85.4 Explain the code
score(chars, match, mismatch):- This function computes the alignment score based on matches and mismatches.
generate_indices(dimensions):- Generates all possible index tuples for alignment, given the sequence lengths.
generate_moves(num_sequences, options):- Generates valid move combinations for alignment, ensuring at least one non-gap move.
add_tuples(u, v):- Element-wise addition of two tuples.
build_scoring_matrix(Strings, score_function):- Constructs the scoring matrix and paths for backtracking.
backtrack_alignment(scoring_matrix, path, Strings):- Reconstructs the alignment based on the scoring matrix and path.
parse_fasta(data):- Parses FASTA format data into a list of sequences.
86 Creating a Restriction Map
For a set \(X\) containing numbers, the difference multiset of \(X\) is the multiset \(ΔX\) defined as the collection of all positive differences between elements of \(X\). As a quick example, if \(X={2,4,7}\), then we will have that \(ΔX={2,3,5}\).
If \(X\) contains n elements, then \(ΔX\) will contain one element for each pair of elements from \(X\), so that \(ΔX\) contains \((n2)\) elements (see combination statistic). You may note the similarity between the difference multiset and the Minkowski difference \(X⊖X\), which contains the elements of \(ΔX\) and their negatives. For the above set \(X\), \(X⊖X\) is \({−5,−3,−2,2,3,5}\).
In practical terms, we can easily obtain a multiset \(L\) corresponding to the distances between restriction sites on a chromosome. If we can find a set \(X\) whose difference multiset \(ΔX\) is equal to \(L\), then \(X\) will represent possible locations of these restriction sites.
Given: A multiset \(L\) containing \((n2)\) positive integers for some positive integer \(n\).
Return: A set \(X\) containing n nonnegative integers such that \(ΔX=L\).
86.1 Sample Dataset
2 2 3 3 4 5 6 7 8 1086.2 Sample Output
0 2 4 7 1086.3 Solution
from math import sqrt
def reconstruct_set(input_data):
"""
Reconstruct the original set from the given differences.
:param input_data: A string containing space-separated integers representing the differences
:return: A list of integers representing the reconstructed set
"""
# Convert input string to list of integers
differences = list(map(int, input_data.strip().split()))
# Calculate the number of elements in the original set
# using the quadratic formula: n(n-1)/2 = len(differences)
set_size = int(0.5 + 0.5 * sqrt(8.0 * len(differences) + 1))
# Initialize the result set with 0
result_set = [0]
# Add the largest difference to the result set
largest_difference = max(differences)
result_set.append(largest_difference)
differences.remove(largest_difference)
# Create a set of unique differences
unique_differences = set(differences)
for candidate in unique_differences:
# Check if the candidate fits with all existing elements in the result set
if sum([(abs(candidate - element) in differences) for element in result_set]) == len(result_set):
for element in result_set:
# Remove the differences we've already accounted for
differences.remove(abs(candidate - element))
# Add the new element to the result set
result_set.append(candidate)
if len(result_set) == set_size:
break
return sorted(result_set)
# Example usage
input_data = """
2 2 3 3 4 5 6 7 8 10
"""
result = reconstruct_set(input_data)
print(' '.join(map(str, result)))86.4 Step-by-Step Explanation
- Convert Input to a List:
- The input string is converted into a list of integers. These integers represent the differences between every pair of elements in the original set.
- Determine the Number of Elements:
- The code calculates how many numbers were in the original set using a mathematical formula related to the number of differences.
- Start with the Smallest Element:
- The code assumes the smallest number in the set is
0and starts theresult_setwith[0].
- The code assumes the smallest number in the set is
- Add the Largest Difference:
- The largest number in the original set is found by taking the largest difference from the list. This number is added to the
result_set.
- The largest number in the original set is found by taking the largest difference from the list. This number is added to the
- Reconstruct the Remaining Numbers:
- The code checks each remaining difference to see if it can be used to find other numbers in the set. It does this by ensuring that each candidate number fits with all previously found numbers (i.e., the differences match).
- Build the Set:
- As valid numbers are found, they are added to the
result_set, and the corresponding differences are removed from the list.
- As valid numbers are found, they are added to the
- Return the Sorted Set:
- The
result_setis sorted and returned, which is the reconstructed original set.
- The
87 Counting Rooted Binary Trees
As in the case of unrooted trees, say that we have a fixed collection of \(n\) taxa labeling the leaves of a rooted binary tree \(T\). You may like to verify that (by extension of “Counting Phylogenetic Ancestors”) such a tree will contain \(n−1\) internal nodes and \(2n−2\) total edges. Any edge will still encode a split of taxa; however, the two splits corresponding to the edges incident to the root of \(T\) will be equal. We still consider two trees to be equivalent if they have the same splits (which requires that they must also share the same duplicated split to be equal).
Let \(B(n)\) represent the total number of distinct rooted binary trees on n labeled taxa.
Given: A positive integer \(n (n≤1000)\).
Return: The value of \(B(n)\) modulo 1,000,000.
87.1 Sample Dataset
487.2 Sample Output
1587.3 Solution
def count_unrooted_binary_trees(n):
'''Returns the number of unrooted binary trees with n leaves.'''
# The total number is just the double factorial (2n - 5)!!
result = 1
for i in range(2 * n - 5, 1, -2):
result = (result * i) % 10**6
return result
def count_rooted_binary_trees(n):
'''Returns the number of rooted binary trees with n leaves.'''
# Can transform an unrooted binary tree into a rooted binary tree by inserting
# a node into any of its 2*n - 3 edges.
return (count_unrooted_binary_trees(n) * (2 * n - 3)) % 10**6
# Read the input data.
input_data = """
4
"""
n = int(input_data.strip())
# Get the number of unrooted binary trees.
count = count_rooted_binary_trees(n)
# Print the answer.
print(count)87.4 count_unrooted_binary_trees(n)
- Purpose: This function calculates the number of possible unrooted binary trees with
nleaves. - Logic:
- The number of unrooted binary trees with
nleaves is given by the double factorial of(2n - 5), which is denoted as(2n - 5)!!. - The double factorial of a number is the product of all integers down to
1that have the same parity (odd/even) as the starting number. - For example, if
n = 4,(2n - 5) = 3, and the double factorial would be3!! = 3. - The loop multiplies all odd numbers from
2n - 5down to3. - The result is taken modulo
10^6to keep the number manageable and avoid overflow.
- The number of unrooted binary trees with
87.5 count_rooted_binary_trees(n)
- Purpose: This function calculates the number of possible rooted binary trees with
nleaves. - Logic:
- A rooted binary tree can be derived from an unrooted binary tree by adding a root to any of the
2n - 3edges of the unrooted tree. - Therefore, the number of rooted binary trees is the number of unrooted binary trees multiplied by
(2n - 3). - Again, the result is taken modulo
10^6.
- A rooted binary tree can be derived from an unrooted binary tree by adding a root to any of the
87.6 How It Works
Input: The code reads the input value
nfrom the stringinput_data. For example, ifn = 4, the code calculates the number of binary trees forn = 4.Execution Flow:
count_rooted_binary_trees(n)is called withn = 4.- Inside this function,
count_unrooted_binary_trees(n)is called. - The
count_unrooted_binary_trees(n)function computes the product(2n - 5)!!modulo10^6:- For
n = 4,(2n - 5) = 3. - The loop runs from
3to1(odd numbers only), resulting in3!! = 3.
- For
- The result (
3in this case) is multiplied by(2 * n - 3) = 5, giving3 * 5 = 15. - The final result is
15 % 10^6 = 15, which is returned and printed.
88 Sex-Linked Inheritance
The conditional probability of an event \(A\) given another event \(B\), written \(Pr(A∣B)\), is equal to \(Pr(A and B)\) divided by \(Pr(B)\).
Note that if \(A\) and \(B\) are independent, then \(Pr(A and B)\) must be equal to \(Pr(A)×Pr(B)\), which results in \(Pr(A∣B)=Pr(A)\). This equation offers an intuitive view of independence: the probability of \(A\), given the occurrence of event \(B\), is simply the probability of \(A\) (which does not depend on \(B\)).
In the context of sex-linked traits, genetic equilibrium requires that the alleles for a gene \(k\) are uniformly distributed over the males and females of a population. In other words, the distribution of alleles is independent of sex.
Given: An array \(A\) of length \(n\) for which \(A[k]\) represents the proportion of males in a population exhibiting the \(k\)-th of \(n\) total recessive X-linked genes. Assume that the population is in genetic equilibrium for all \(n\) genes.
Return: An array \(B\) of length \(n\) in which \(B[k]\) equals the probability that a randomly selected female will be a carrier for the \(k\)-th gene.
88.1 Sample Dataset
0.1 0.5 0.888.2 Sample Output
0.18 0.5 0.3288.3 Solution
# Read the input data.
input_data = """
0.1 0.5 0.8
"""
# Convert input data to a list of floats.
numbers = [float(x) for x in input_data.strip().split()]
# Calculate the desired values.
results = [2 * (x - x**2) for x in numbers]
# Format the results to two decimal places and print.
formatted_results = ' '.join(f"{result:f}" for result in results)
print(formatted_results)
unformatted_results = ' '.join(f"{result}" for result in results)
print(unformatted_results) # only unformatted_results accepted to answerCalculate Results: - For each number in numbers, the code calculates a new value using the formula 2 * (x - x**2). This formula computes the difference between a number and its square, doubles it, and stores it in the results list. - The calculations for each number would be: - For 0.1: 2 * (0.1 - 0.1**2) = 0.18 - For 0.5: 2 * (0.5 - 0.5**2) = 0.50 - For 0.8: 2 * (0.8 - 0.8**2) = 0.32 - The resulting list is: [0.18, 0.50, 0.32].
89 Phylogeny Comparison with Split Distance
Define the split distance between two unrooted binary trees as the number of nontrivial splits contained in one tree but not the other.
Formally, if \(s(T1,T2)\) denotes the number of nontrivial splits shared by unrooted binary trees \(T1\) and \(T2\), Then their split distance is \(d_{split}(T1,T2)=2(n−3)−2s(T1,T2)\).
Given: A collection of at most 3,000 species taxa and two unrooted binary trees \(T1\) and \(T2\)on these taxa in Newick format.
Return: The split distance \(d_{split}(T1,T2)\).
89.1 Sample Dataset
dog rat elephant mouse cat rabbit
(rat,(dog,cat),(rabbit,(elephant,mouse)));
(rat,(cat,dog),(elephant,(mouse,rabbit)));89.2 Sample Output
289.3 Solution
import random
def get_fingerprints_list(taxa_dict, tree):
result = []
last_char = ''
taxon = ''
taxa_stack = []
for char in tree:
if char in ('(', ',', ')'):
if last_char in ('(', ','):
if taxon:
taxa_stack.append(taxa_dict[taxon])
taxon = ''
elif last_char == ')':
t1 = taxa_stack.pop()
t2 = taxa_stack.pop()
result.append(t1 ^ t2)
taxa_stack.append(t1 ^ t2)
last_char = char
else:
if char != ' ':
taxon += char
return result
def find_split_distance(taxa, tree1, tree2):
random.seed() # Initialize random number generator
taxa_dict = {taxon: random.randint(0, 2**12) for taxon in taxa} # Adjusted bit range for randomness
fingerprints1 = sorted(get_fingerprints_list(taxa_dict, tree1))
fingerprints2 = sorted(get_fingerprints_list(taxa_dict, tree2))
shared_count = 0
i, j = len(fingerprints1) - 1, len(fingerprints2) - 1
while i >= 0 and j >= 0:
if fingerprints1[i] == fingerprints2[j]:
shared_count += 1
i -= 1
j -= 1
elif fingerprints1[i] > fingerprints2[j]:
i -= 1
else:
j -= 1
return 2 * (len(taxa) - 3) - 2 * shared_count
# Input data
sample_input = """
dog rat elephant mouse cat rabbit
(rat,(dog,cat),(rabbit,(elephant,mouse)));
(rat,(cat,dog),(elephant,(mouse,rabbit)));
"""
input_lines = sample_input.strip().split("\n")
taxa = input_lines[0].split()
tree1 = input_lines[1]
tree2 = input_lines[2]
# Compute the maximum split distance over 500 iterations
max_distance = max(find_split_distance(taxa, tree1, tree2) for _ in range(500))
print(max_distance)89.4 Explanation
- Function
get_fingerprints_list(taxa_dict, tree):- Purpose: Converts a tree in Newick format into a list of fingerprints based on a dictionary of taxon identifiers.
- How It Works:
- Iterates through characters in the tree string.
- Handles tree structure symbols
(,,,)and taxon names. - Uses a stack (
taxa_stack) to keep track of taxon fingerprints. - When encountering
), it combines the fingerprints of the last two taxa in the stack using the XOR operation (^), which is a common way to handle such trees.
- Function
find_split_distance(taxa, tree1, tree2):- Purpose: Computes the split distance between two trees.
- How It Works:
- Generates a random dictionary mapping taxa to unique integer fingerprints.
- Calculates fingerprints for both trees and sorts them.
- Finds the number of shared fingerprints between the two trees.
- Computes the split distance using the formula
2 * (n - 3) - 2 * shared_count, wherenis the number of taxa.
- Main Execution:
- Input Handling: Reads and parses input data.
- Computation: Runs the
find_split_distancefunction 500 times with random initialization to determine the maximum split distance. - Output: Prints the maximum split distance found.
90 The Wright-Fisher Model of Genetic Drift
Consider flipping a weighted coin that gives “heads” with some fixed probability pp (i.e., \(p\) is not necessarily equal to 1/2).
We generalize the notion of binomial random variable from “Independent Segregation of Chromosomes” to quantify the sum of the weighted coin flips. Such a random variable \(X\) takes a value of \(k\) if a sequence of \(n\) independent “weighted coin flips” yields \(k\) “heads” and \(n−k\) “tails.” We write that \(X∈Bin(n,p)\).
To quantify the Wright-Fisher Model of genetic drift, consider a population of \(N\) diploid individuals, whose \(2N\) chromosomes posses mm copies of the dominant allele. As in “Counting Disease Carriers”, set \(p= \frac{m}{2N}\). Next, recall that the next generation must contain exactly \(N\) individuals. These individuals’ \(2N\) alleles are selected independently: a dominant allele is chosen with probability pp, and a recessive allele is chosen with probability \(1−p\).
Given: Positive integers \(N\) (\(N≤7\)), \(m\) (\(m≤2N\)), \(g\) (\(g≤6\)) and \(k\) (\(k≤2N\)).
Return: The probability that in a population of \(N\) diploid individuals initially possessing mm copies of a dominant allele, we will observe after \(g\) generations at least \(k\) copies of a recessive allele. Assume the Wright-Fisher model.
90.1 Sample Dataset
4 6 2 190.2 Sample Output
0.77290.3 Solution
from scipy.special import comb
def calculate_initial_probabilities(N, m):
"""
Calculate the probabilities of having a given number of recessive alleles in the first generation.
"""
p_rec = 1 - m / (2.0 * N)
return [comb(2 * N, i) * p_rec ** i * (1 - p_rec) ** (2 * N - i) for i in range(1, 2 * N + 1)]
def update_probabilities(previous_p, N):
"""
Update the probabilities of recessive alleles for the next generation.
"""
new_p = []
for j in range(1, 2 * N + 1):
temp = [comb(2 * N, j) * (x / (2 * N)) ** j * (1 - x / (2 * N)) ** (2 * N - j) for x in range(1, 2 * N + 1)]
new_p.append(sum(temp[i] * previous_p[i] for i in range(len(temp))))
return new_p
def calculate_final_probability(N, m, g, k):
"""
Calculate the probability of observing at least k recessive alleles after g generations.
"""
previous_p = calculate_initial_probabilities(N, m)
for _ in range(2, g + 1):
previous_p = update_probabilities(previous_p, N)
return sum(previous_p[k - 1:])
# Sample input
sample_input = """
4 6 2 1
"""
input_lines = sample_input.strip().split("\n")
N, m, g, k = [int(x) for x in input_lines[0].split()]
# Calculate and print the final probability
final_prob = calculate_final_probability(N, m, g, k)
print(final_prob)90.4 Explanation
- Function
calculate_initial_probabilities(N, m):- Purpose: Computes the probabilities of having different numbers of recessive alleles in the first generation.
- How It Works: Uses the
combfunction to calculate binomial probabilities based on the initial proportion of recessive alleles.
- Function
update_probabilities(previous_p, N):- Purpose: Updates the probabilities for subsequent generations.
- How It Works: For each possible number of recessive alleles, calculates the new probabilities based on the previous generation’s probabilities and the binomial distribution.
- Function
calculate_final_probability(N, m, g, k):- Purpose: Computes the final probability of observing at least
krecessive alleles afterggenerations. - How It Works: Iterates through generations, updating probabilities each time. After
ggenerations, it sums up the probabilities for having at leastkrecessive alleles.
- Purpose: Computes the final probability of observing at least
- Main Execution:
- Input Handling: Reads and parses input data.
- Computation: Uses the functions to calculate the final probability.
- Output: Prints the result.
91 Alignment-Based Phylogeny
Say that we have \(n\) taxa represented by strings \(s_1,s_2,…,s_n\) with a multiple alignment inducing corresponding augmented strings \(\hat s_1, \hat s_2,…, \hat s_n\).
Recall that the number of single-symbol substitutions required to transform one string into another is the Hamming distance between the strings (see “Counting Point Mutations”). Say that we have a rooted binary tree \(T\) containing \(\hat s_1, \hat s_2,…, \hat s_n\) at its leaves and additional strings \(\hat s_{n+1}, \hat s_{n+2),…, \hat s_{2n-1}\). at its internal nodes, including the root (the number of internal nodes is \(n−1\) by extension of “Counting Phylogenetic Ancestors”). Define \(d_H(T)\) as the sum of \(dH(\hat s_i, \hat s_j)\) over all edges \((\hat s_i, \hat s_j)\) in \(T\):
\[d_H(T)= \sum _{{ \hat s_i, \hat s_j} ∈E (T)} dH(\hat s_i, \hat s_j)\]
Thus, our aim is to minimize \(d_H(T)\).
Given: A rooted binary tree \(T\) on \(n\) (\(n≤500\)) species, given in Newick format, followed by a multiple alignment of \(m\) (\(m≤n\)) augmented DNA strings having the same length (at most 300 bp) corresponding to the species and given in FASTA format.
Return: The minimum possible value of \(dH(T)\), followed by a collection of DNA strings to be assigned to the internal nodes of \(T\) that will minimize \(dH(T)\) (multiple solutions will exist, but you need only output one).
91.1 Sample Dataset
(((ostrich,cat)rat,(duck,fly)mouse)dog,(elephant,pikachu)hamster)robot;
>ostrich
AC
>cat
CA
>duck
T-
>fly
GC
>elephant
-T
>pikachu
AA91.2 Sample Output
8
>rat
AC
>mouse
TC
>dog
AC
>hamster
AT
>robot
AC91.3 Solution
from math import inf
import re
from collections import defaultdict
def parse_newick(newick, directed=True):
newick = re.sub(",,", ",.,", newick)
newick = re.sub(r"\(,", "(.,", newick)
newick = re.sub(r",\)", ",.)", newick)
newick = re.sub(r"\(\)", "(.)", newick)
newick = re.sub(r"^\((.+)\);", r"\1", newick)
m = re.finditer(r"(\(|[A-z_.]+|,|\))", newick)
tokens = [x.group() for x in m]
count = 0
node_stack = ["0"]
g = defaultdict(list)
i = len(tokens) - 1
while i >= 0:
if tokens[i] == "(":
node_stack = node_stack[:-1]
elif tokens[i] == ")":
if i + 1 < len(tokens) and tokens[i + 1] not in ",)":
node = tokens[i + 1]
else:
count += 1
node = str(count)
g[node_stack[-1]].append({"n": node, "w": 1})
if not directed:
g[node].append({"n": node_stack[-1], "w": 1})
node_stack += [node]
elif tokens[i] != "," and (i == 0 or tokens[i - 1] != ")"):
if tokens[i] == ".":
count += 1
tokens[i] = str(count)
g[node_stack[-1]].append({"n": tokens[i], "w": 1})
if not directed:
g[tokens[i]].append({"n": node_stack[-1], "w": 1})
i -= 1
return g
clas Rec:
"""A simple FASTA record"""
def __init__(self, id, seq):
self.id = id
self.seq = seq
def __len__(self):
return len(self.seq)
def read_fasta(input_string):
lines = input_string.strip().split('\n')
header, sequence = "", []
for line in lines:
if line.startswith(">"):
if sequence:
yield Rec(header, "".join(sequence))
header, sequence = line[1:], []
elif line.strip(): # 빈 줄 무시
sequence.append(line.strip())
if sequence:
yield Rec(header, "".join(sequence))
def nodes(graph):
s = list(graph.keys())
e = [y for v in graph.values() for y in v]
return set(s) | set(e)
# return all leaves of a simple graph
def leaves(graph):
return nodes(graph) - set(graph.keys())
def extract_position(graph, seqs, pos):
chars = {}
for n in nodes(graph) - leaves(graph):
chars[n] = ""
for leaf in leaves(graph):
chars[leaf] = seqs[leaf][pos]
return chars
def traceback(skp, node, ind):
bases = ["A", "C", "T", "G", "-"]
chars = {}
chars[node] = bases[ind]
for k, v in skp[node][ind].items():
if k in skp:
chars = chars | traceback(skp, k, v)
return chars
def small_parsimony(graph, chars):
bases = ["A", "C", "T", "G", "-"]
sk = {} # minimum parsimony score of the subtree over possible labels
skp = {} # pointer to selected base for each child over possible labels
to_proces = nodes(graph)
# # initialise leaves
for leaf in leaves(graph):
sk[leaf] = [0 if chars[leaf] == c else inf for c in bases]
to_process.remove(leaf)
# iterate over available nodes till all are processed
while to_process:
for n in list(to_process):
if all(v in sk for v in graph[n]):
sk[n], skp[n] = [], []
for k in bases:
tot = 0
ptr = {}
for d, sk_child in [(d, sk[d]) for d in graph[n]]:
score = []
for i, c in enumerate(bases):
score += [sk_child[i] + (0 if c == k else 1)]
tot += min(score)
ptr[d] = score.index(min(score))
skp[n] += [ptr]
sk[n] += [tot]
to_process.remove(n)
# Recover sequence
node = "0"
score = min(sk[node])
return score, traceback(skp, node, sk[node].index(score))
def alph(tree, seqs, i):
# initialise sequences
for n in nodes(tree) - leaves(tree):
seqs[n] = ""
n = len(seqs[list(leaves(tree))[0]])
total_score = 0
for pos in range(n):
chars = extract_position(tree, seqs, pos)
score, tbchars = small_parsimony(tree, chars)
total_score += score
for k, v in tbchars.items():
seqs[k] += v
return total_score, seqs
def simplify_tree(graph):
return {k: [x["n"] for x in v] for k, v in graph.items()}
sample_input = """
(((ostrich,cat)rat,(duck,fly)mouse)dog,(elephant,pikachu)hamster)robot;
>ostrich
AC
>cat
CA
>duck
T-
>fly
GC
>elephant
-T
>pikachu
AA
"""
tree = parse_newick(sample_input.strip().split('\n')[0])
tree = simplify_tree(tree)
seqs = read_fasta('\n'.join(sample_input.strip().split('\n')[1:]))
seqs = {x.id: x.seq for x in seqs}
total_score, seqs = alph(tree, seqs, 1)
print(total_score)
for node in tree.keys():
if node != "0":
print(f">{node}")
print(seqs[node])91.4 Working Principle
The code implements the Small Parsimony algorithm for phylogenetic tree reconstruction.
It starts by parsing a Newick format string representation of a tree using the
parse_newickfunction. This function creates a graph representation of the tree.The
small_parsimonyfunction is the core of the algorithm. It calculates the most parsimonious ancestral sequences for internal nodes of the tree.The algorithm works bottom-up, starting from the leaves and moving towards the root:
- For leaves, it initializes scores based on their known character states.
- For internal nodes, it calculates scores for each possible base by considering the scores of its children.
The
tracebackfunction is used to reconstruct the most parsimonious ancestral sequences by traversing the tree from root to leaves.The
alphfunction applies the Small Parsimony algorithm to each position in the sequences, building up the full ancestral sequences.Finally, the code reads a sample input (in FASTA format), constructs the tree, applies the algorithm, and prints the results.
This algorithm aims to find the ancestral sequences that minimize the total number of mutations (changes) along the branches of the phylogenetic tree, based on the principle of maximum parsimony.
92 Assessing Assembly Quality with N50 and N75
Given a collection of DNA strings representing contigs, we use the N statistic NXX (where XX ranges from 01 to 99) to represent the maximum positive integer \(L\) such that the total number of nucleotides of all contigs having length \(≥L\) is at least XX% of the sum of contig lengths. The most commonly used such statistic is N50, although N75 is also worth mentioning.
Given: A collection of at most 1000 DNA strings (whose combined length does not exceed 50 kbp).
Return: N50 and N75 for this collection of strings.
92.1 Sample Dataset
GATTACA
TACTACTAC
ATTGAT
GAAGA92.2 Sample Output
7 692.3 Solution
def calculate_nxx(contigs, xx):
total_length = sum(len(contig) for contig in contigs)
target_length = total_length * xx / 100
sorted_contigs = sorted(contigs, key=len, reverse=True)
cumulative_length = 0
for contig in sorted_contigs:
cumulative_length += len(contig)
if cumulative_length >= target_length:
return len(contig)
return 0
# Read input
sample_input = """
GATTACA
TACTACTAC
ATTGAT
GAAGA
"""
contigs = [line.strip() for line in sample_input.strip().split("\n")]
# Calculate N50 and N75
n50 = calculate_nxx(contigs, 50)
n75 = calculate_nxx(contigs, 75)
# Print results
print(f"{n50} {n75}")The code calculates N50 and N75 values, which are measures used to asses the quality of DNA sequence assemblies.
92.4 Breaking Down the Steps:
calculate_nxx(contigs, xx)Function:- Input: A list of DNA sequences (
contigs) and a percentage (xxlike 50 for N50). - Output: The length of the sequence (contig) where the cumulative length reaches the specified percentage of the total length.
- How It Works:
- Step 1: Add up the lengths of all sequences to get the total length.
- Step 2: Sort the sequences from longest to shortest.
- Step 3: Add lengths one by one from the sorted list until the sum reaches the specified percentage of the total length. The length of the last added sequence is the NXX value.
- Input: A list of DNA sequences (
- Main Code:
- The sample input is split into individual DNA sequences.
- The code then calculates:
- N50: The sequence length where 50% of the total length is reached.
- N75: The sequence length where 75% of the total length is reached.
- Finally, it prints these N50 and N75 values.
93 Fixing an Inconsistent Character Set
A submatrix of a matrix \(M\) is a matrix formed by selecting rows and columns from \(M\) and taking only those entries found at the intersections of the selected rows and columns. We may also think of a submatrix as formed by deleting the remaining rows and columns from \(M\).
Given: An inconsistent character table \(C\) on at most 100 taxa.
Return: A submatrix of \(C′\) representing a consistent character table on the same taxa and formed by deleting a single row of \(C\). (If multiple solutions exist, you may return any one.)
93.1 Sample Dataset
100001
000110
111000
10011193.2 Sample Output
000110
100001
10011193.3 Solution
from collections import defaultdict
def conflict(c1, c2):
# 모든 인덱스에 대해 한 번에 비교하여 충돌 여부 확인
return any((c1[i] == 1 and c2[i] == 0) or (c1[i] == 0 and c2[i] == 1) for i in range(len(c1)))
def conflicts(characters):
count = defaultdict(int)
for i in range(len(characters)):
for j in range(i + 1, len(characters)):
if conflict(characters[i], characters[j]):
count[i] += 1
count[j] += 1
return count
# 입력 처리
sample_input = """
100001
000110
111000
100111
"""
lines = sample_input.strip().split("\n")
characters = [[int(x) for x in ch] for ch in lines]
# 충돌 계산
count = conflicts(characters)
# 가장 많은 충돌을 가진 행 제거
rm = max(count, key=count.get)
# 결과 출력
print(*lines[:rm], *lines[rm + 1:], sep="\n")The code identifies and removes the row from a set of binary sequences (like 100001) that has the most conflicts with other rows. A “conflict” is defined as one sequence having a 1 where another has a 0, and vice versa.
93.4 Breaking Down the Steps:
conflict(c1, c2)Function:- Purpose: Check if two sequences (
c1andc2) conflict with each other. - How It Works:
- It compares the two sequences at each index.
- If at any index, one sequence has
1and the other has0, they are in conflict. - The function returns
Trueif there is any conflict; otherwise, it returnsFalse.
- Purpose: Check if two sequences (
conflicts(characters)Function:- Purpose: Count how many conflicts each sequence has with all other sequences.
- How It Works:
- It compares each sequence with every other sequence.
- If two sequences conflict, it increments a conflict counter for both sequences.
- It returns a dictionary where the key is the sequence index, and the value is the number of conflicts that sequence has.
- Main Code:
- Input Processing:
- The binary sequences are read and converted into lists of integers.
- Conflict Counting:
- The code uses the
conflictsfunction to count how many conflicts each sequence has.
- The code uses the
- Removing the Most Conflicting Sequence:
- The sequence with the highest number of conflicts is identified.
- This sequence is then removed from the list.
- Output:
- The remaining sequences (with the most conflicting one removed) are printed.
- Input Processing:
94 Wright-Fisher’s Expected Behavior
In “The Wright-Fisher Model of Genetic Drift”, we generalized the concept of a binomial random variable \(Bin(n,p)\) as a “weighted coin flip.” It is only natural to calculate the expected value of such a random variable.
For example, in the case of unweighted coin flips (i.e., \(p=1/2\)), our intuition would indicate that \(E(Bin(n/2))\) is \(n/2\); what should be the expected value of a binomial random variable?
Given: A positive integer \(n\) (\(n≤1000000\)) followed by an array \(P\) of length mm (\(m≤20\)) containing numbers between 0 and 1. Each element of \(P\) can be seen as representing a probability corresponding to an allele frequency.
Return: An array \(B\) of length mm for which \(B[k]\) is the expected value of \(Bin(n,P[k])\); in terms of Wright-Fisher, it represents the expected allele frequency of the next generation.
94.1 Sample Dataset
17
0.1 0.2 0.394.2 Sample Output
1.7 3.4 5.194.3 Solution
The problem asks us to calculate the expected value of a binomial random variable given a population size \(n\) and an array \(P\) of allele frequencies. For each element \(p\) in the array \(P\), we need to compute the expected value \(E(Bin(n, p))\).
For a binomial random variable \(Bin(n, p)\), where: - \(n\) is the number of trials (in this case, the number of individuals in the population), - \(p\) is the probability of succes (or the allele frequency),
The expected value \(E(Bin(n, p))\) is calculated as:
[ E(Bin(n, p)) = n p ]
Given that, the task is to compute this value for each probability in the array \(P\).We can implement this in Python as follows:
# Sample Input
sample_input = """
17
0.1 0.2 0.3
"""
# Parse input
lines = sample_input.strip().split('\n')
n = int(lines[0])
P = list(map(float, lines[1].split()))
# Calculate the expected values
B = [n * p for p in P]
# Print the result
print(' '.join(map(str, B)))94.4 Explanation
- Input Parsing: We read the population size \(n\) and the array \(P\) of allele frequencies.
- Computation: For each \(p\) in \(P\), we compute \(n \times p\) and store it in array \(B\).
- Output: Finally, we print the values in \(B\) as a space-separated string.
95 The Founder Effect and Genetic Drift
Given: Two positive integers \(N\) and \(m\), followed by an array \(A\) containing \(k\) integers between 0 and \(2N\). \(A[j]\) represents the number of recessive alleles for the \(j\)-th factor in a population of \(N\) diploid individuals.
Return: An \(m×k\) matrix \(B\) for which \(B_{i,j}\) represents the common logarithm of the probability that after \(i\) generations, no copies of the recessive allele for the \(j\)-th factor will remain in the population. Apply the Wright-Fisher model.
95.1 Sample Dataset
4 3
0 1 295.2 Sample Output
0.0 -0.463935575821 -0.999509892866
0.0 -0.301424998891 -0.641668367342
0.0 -0.229066698008 -0.48579855245695.3 Solution
from scipy.special import comb
import numpy as np
def wright_fisher_genetic_drift(N, m, g):
"""
Calculate the log10 of the probability that no copies of the recessive allele remain in the population
after g generations, given an initial count of m recessive alleles.
"""
q = m / (2 * N) # Initial dominant allele frequency
p = 1 - q # Initial recessive allele frequency
# Initialize probability of exactly t recessive alleles in the first generation
prob = np.array([comb(2 * N, i) * (q ** i) * (p ** (2 * N - i)) for i in range(1, 2 * N + 1)])
# Iterate through generations
for _ in range(1, g):
# Calculate probabilities for the next generation
next_prob = np.zeros(2 * N)
for t in range(1, 2 * N + 1):
# Calculate probability of having exactly t recessive alleles in the current generation
prob_t = np.array([comb(2 * N, t) * ((i / (2 * N)) ** t) * ((1 - (i / (2 * N))) ** (2 * N - t)) for i in range(1, 2 * N + 1)])
next_prob[t - 1] = np.sum(prob_t * prob)
prob = next_prob
# Return the log10 of the probability of no recessive alleles remaining
return np.log10(1 - np.sum(prob))
def calculate_genetic_drift_matrix(N, m, A):
"""
Generate the matrix B where B[i, j] represents the common logarithm of the probability that after i generations,
no copies of the recessive allele for the j-th factor will remain in the population.
"""
k = len(A)
B = np.zeros((m, k))
for i in range(m):
for j in range(k):
B[i, j] = wright_fisher_genetic_drift(N, A[j], i + 1)
return B
# Sample Input
sample_input = """
4 3
0 1 2
"""
# Parse input
lines = sample_input.strip().split('\n')
N, m = map(int, lines[0].split())
A = list(map(int, lines[1].split()))
# Calculate the matrix B
B = calculate_genetic_drift_matrix(N, m, A)
# Print the results
for row in B:
print(' '.join(map(str, row)))This code calculates the probability of losing all copies of a recessive allele in a population over several generations, based on the Wright-Fisher model of genetic drift. The result is a matrix where each entry tells us the likelihood (in logarithmic form) that no recessive alleles remain after a given number of generations.
95.4 Functions and Their Roles
wright_fisher_genetic_drift(N, m, g)Purpose: Calculates the probability of losing all recessive alleles after
ggenerations, starting withmrecessive alleles in a population of sizeN.How It Works:
- Initial Setup:
qis the frequency of dominant alleles.pis the frequency of recessive alleles.
- First Generation:
- Calculate the probability of having exactly
trecessive alleles in the first generation.
- Calculate the probability of having exactly
- Subsequent Generations:
- For each generation, update the probabilities based on the previous generation.
- Final Calculation:
- Compute the probability that no recessive alleles remain after
ggenerations and return its log base 10.
- Compute the probability that no recessive alleles remain after
- Initial Setup:
calculate_genetic_drift_matrix(N, m, A)Purpose: Creates a matrix where each entry shows the log probability that no recessive alleles remain after a certain number of generations for various initial counts of recessive alleles.
How It Works:
- Matrix Initialization:
Bis initialized as a zero matrix.
- Filling the Matrix:
- For each possible number of generations and each initial count of recessive alleles, calculate the log probability using
wright_fisher_genetic_driftand store it in the matrixB.
- For each possible number of generations and each initial count of recessive alleles, calculate the log probability using
- Matrix Initialization:
95.5 Key Points
wright_fisher_genetic_driftfocuses on updating probabilities generation by generation.calculate_genetic_drift_matrixbuilds a matrix from these probabilities for different scenarios.
96 Global Alignment with Scoring Matrix and Affine Gap Penalty
An affine gap penalty is written as \(a+b⋅(L−1)\), where \(L\) is the length of the gap, aa is a positive constant called the gap opening penalty, and \(b\) is a positive constant called the gap extension penalty.
We can view the gap opening penalty as charging for the first gap symbol, and the gap extension penalty as charging for each subsequent symbol added to the gap.
For example, if \(a=11\) and \(b=1\), then a gap of length 1 would be penalized by 11 (for an average cost of 11 per gap symbol), whereas a gap of length 100 would have a score of 110 (for an average cost of 1.10 per gap symbol).
Consider the strings “PRTEINS” and “PRTWPSEIN”. If we use the BLOSUM62 scoring matrix and an affine gap penalty with \(a=11\) and \(b=1\), then we obtain the following optimal alignment.
PRT---EINS
||| |||
PRTWPSEIN-Matched symbols contribute a total of 32 to the calculation of the alignment’s score, and the gaps cost 13 and 11 respectively, yielding a total score of 8.
Given: Two protein strings \(s\) and \(t\) in FASTA format (each of length at most 100 aa).
Return: The maximum alignment score between \(s\) and \(t\), followed by two augmented strings \(s′\) and \(t′\) representing an optimal alignment of \(s\) and \(t\). Use:
- The BLOSUM62 scoring matrix.
- Gap opening penalty equal to 11.
- Gap extension penalty equal to 1.
96.1 Sample Dataset
>Rosalind_49
PRTEINS
>Rosalind_47
PRTWPSEIN96.2 Sample Output
8
PRT---EINS
PRTWPSEIN-96.3 Solution
# BLOSUM62 matrix as a string
blosum62_str = """
A C D E F G H I K L M N P Q R S T V W Y
A 4 0 -2 -1 -2 0 -2 -1 -1 -1 -1 -2 -1 -1 -1 1 0 0 -3 -2
C 0 9 -3 -4 -2 -3 -3 -1 -3 -1 -1 -3 -3 -3 -3 -1 -1 -1 -2 -2
D -2 -3 6 2 -3 -1 -1 -3 -1 -4 -3 1 -1 0 -2 0 -1 -3 -4 -3
E -1 -4 2 5 -3 -2 0 -3 1 -2 -2 0 -1 2 0 0 -1 -2 -3 -2
F -2 -2 -3 -3 6 -3 -1 0 -3 0 0 -3 -4 -3 -3 -2 -2 -1 1 3
G 0 -3 -1 -2 -3 6 -2 -4 -2 -4 -3 0 -2 -2 -3 0 -2 -3 -2 -3
H -2 -3 -1 0 -1 -2 8 -3 -1 -3 -2 1 -2 0 0 -1 -2 -3 -2 2
I -1 -1 -3 -3 0 -4 -3 4 -3 2 1 -3 -3 -3 -3 -2 -1 3 -3 -1
K -1 -3 -1 1 -3 -2 -1 -3 5 -2 -1 0 -1 1 2 0 -1 -2 -3 -2
L -1 -1 -4 -2 0 -4 -3 2 -2 4 2 -3 -3 -2 -2 -2 -1 1 -2 -1
M -1 -1 -3 -2 0 -3 -2 1 -1 2 5 -2 -2 0 -1 -1 -1 1 -1 -1
N -2 -3 1 0 -3 0 1 -3 0 -3 -2 6 -2 0 0 1 0 -3 -4 -2
P -1 -3 -1 -1 -4 -2 -2 -3 -1 -3 -2 -2 7 -1 -2 -1 -1 -3 -4 -3
Q -1 -3 0 2 -3 -2 0 -3 1 -2 0 0 -1 5 1 0 -1 -2 -2 -1
R -1 -3 -2 0 -3 -3 0 -3 2 -2 -1 0 -2 1 5 -1 -1 -3 -3 -2
S 1 -1 0 0 -2 0 -1 -2 0 -2 -1 1 -1 0 -1 4 1 -2 -3 -2
T 0 -1 -1 -1 -2 -2 -2 -1 -1 -1 -1 0 -1 -1 -1 1 5 0 -2 -2
V 0 -1 -3 -2 -1 -3 -3 3 -2 1 1 -3 -3 -2 -3 -2 0 4 -3 -1
W -3 -2 -4 -3 1 -2 -2 -3 -3 -2 -1 -4 -4 -2 -3 -3 -2 -3 11 2
Y -2 -2 -3 -2 3 -3 2 -1 -2 -1 -1 -2 -3 -1 -2 -2 -2 -1 2 7
"""
def parse_blosum62(matrix_str):
"""Parse the BLOSUM62 matrix from a string into a dictionary."""
lines = matrix_str.strip().split('\n')
headers = lines[0].split()
matrix = {}
for line in lines[1:]:
values = line.split()
row = values[0]
scores = list(map(int, values[1:]))
matrix.update({(row, col): score for col, score in zip(headers, scores)})
return matrix
def parse_fasta(data):
"""Parse FASTA format data into a list of sequences."""
sequences = []
seq = ""
for line in data.strip().split('\n'):
if line.startswith('>'):
if seq:
sequences.append(seq)
seq = ""
else:
seq += line.strip()
if seq:
sequences.append(seq)
return sequences
def match_score(scoring_matrix, a, b):
"""Return the score from the scoring matrix, defaulting to 0 if not found."""
return scoring_matrix.get((a, b), 0)
def global_align_with_affine(s, t, scores, gap, gap_e):
"""Perform global alignment with affine gap penalties."""
m, n = len(s), len(t)
# Initialize matrices
M = [[0] * (n + 1) for _ in range(m + 1)]
X = [[0] * (n + 1) for _ in range(m + 1)]
Y = [[0] * (n + 1) for _ in range(m + 1)]
traceM = [[0] * (n + 1) for _ in range(m + 1)]
traceX = [[0] * (n + 1) for _ in range(m + 1)]
traceY = [[0] * (n + 1) for _ in range(m + 1)]
# Initialize edges
for i in range(1, m + 1):
M[i][0] = gap + gap_e * (i - 1)
X[i][0] = Y[i][0] = float('-inf')
for j in range(1, n + 1):
M[0][j] = gap + gap_e * (j - 1)
X[0][j] = Y[0][j] = float('-inf')
# Fill matrices
for i in range(1, m + 1):
for j in range(1, n + 1):
costX = [M[i-1][j] + gap, X[i-1][j] + gap_e]
X[i][j] = max(costX)
traceX[i][j] = costX.index(X[i][j])
costY = [M[i][j-1] + gap, Y[i][j-1] + gap_e]
Y[i][j] = max(costY)
traceY[i][j] = costY.index(Y[i][j])
costM = [M[i-1][j-1] + match_score(scores, s[i-1], t[j-1]), X[i][j], Y[i][j]]
M[i][j] = max(costM)
traceM[i][j] = costM.index(M[i][j])
# Get maximum score and initialize aligned strings
max_score = M[m][n]
s_align, t_align = s, t
# Traceback
i, j = m, n
while i > 0 or j > 0:
traceback = max([(X[i][j], 0), (Y[i][j], 1), (M[i][j], 2)], key=lambda x: x[0])[1]
if traceback == 0:
t_align = t_align[:j] + '-' + t_align[j:]
i -= 1
elif traceback == 1:
s_align = s_align[:i] + '-' + s_align[i:]
j -= 1
elif traceback == 2:
if traceM[i][j] == 0:
traceback = 0
elif traceM[i][j] == 1:
traceback = 1
i -= 1
j -= 1
# Handle leading gaps
s_align = '-' * j + s_align
t_align = '-' * i + t_align
return str(max_score), s_align, t_align
# Sample dataset in FASTA format
sample_input = """
>Rosalind_49
PRTEINS
>Rosalind_47
PRTWPSEIN
"""
# Parse the FASTA input to get the sequences
sequences = parse_fasta(sample_input)
s, t = sequences[0], sequences[1]
# Parse the BLOSUM62 matrix
blosum62 = parse_blosum62(blosum62_str)
# Perform global alignment with affine gap penalties
alignment = global_align_with_affine(s, t, blosum62, -11, -1)
print('\n'.join(alignment))This Python code performs global sequence alignment between two protein sequences using the BLOSUM62 substitution matrix and affine gap penalties. The alignment proces is a common technique in bioinformatics to compare two sequences and find the best way to align them by inserting gaps and matching characters.
96.4 Detailed Explanation
- BLOSUM62 Matrix as a String:
- The BLOSUM62 matrix, a commonly used substitution matrix in bioinformatics, is provided as a multiline string. It contains scores representing how likely it is for each amino acid pair to substitute for each other.
parse_blosum62(matrix_str)Function:- Purpose: Converts the BLOSUM62 string into a dictionary for easier lookup.
- How It Works:
- The string is split into lines and then into individual elements.
- The first line contains the amino acid headers.
- Each subsequent line contains scores for substituting one amino acid with others.
- A dictionary is created where each key is a tuple of two amino acids, and the value is the corresponding substitution score.
parse_fasta(data)Function:- Purpose: Parses sequences from the FASTA format, which is a standard text-based format for representing sequences.
- How It Works:
- The function reads the input line by line.
- Lines starting with
>indicate sequence headers, which are ignored. - Sequence data is collected into a list of sequences.
match_score(scoring_matrix, a, b)Function:- Purpose: Retrieves the substitution score for a pair of amino acids from the BLOSUM62 matrix.
- How It Works:
- It looks up the score for the amino acid pair
(a, b)in the dictionary. If the pair is not found, it returns0.
- It looks up the score for the amino acid pair
global_align_with_affine(s, t, scores, gap, gap_e)Function:- Purpose: Performs global alignment of two sequences using affine gap penalties.
- How It Works:
- Initialization: Three matrices (
M,X,Y) are used to keep track of the scores for matches, gaps in one sequence, and gaps in the other sequence, respectively.traceM,traceX, andtraceYtrack the path for traceback. - Matrix Filling: The matrices are filled in a nested loop:
M[i][j]: Maximum score considering a match or mismatch.X[i][j]: Maximum score considering a gap in sequencet.Y[i][j]: Maximum score considering a gap in sequences.
- Traceback: After filling the matrices, the function traces back from the last cell to reconstruct the aligned sequences, inserting gaps where needed.
- The traceback ensures that the sequences are aligned optimally according to the scoring matrix and gap penalties.
- Initialization: Three matrices (
- Sample Input and Execution:
- Sample Input: Two sequences (
PRTEINSandPRTWPSEIN) are provided in FASTA format. - Execution:
- The sequences are parsed from the input.
- The BLOSUM62 matrix is parsed.
- Global alignment with affine gap penalties is performed using the parsed sequences and matrix.
- The alignment result, including the score and the aligned sequences, is printed.
- Sample Input: Two sequences (
97 Genome Assembly with Perfect Coverage and Repeats
Recall that a directed cycle is a cycle in a directed graph in which the head of one edge is equal to the tail of the following edge.
In a de Bruijn graph of k-mers, a circular string s is constructed from a directed cycle \(s1→s2→...→si→s1\) is given by \(s1+s2[k]+...+s_{i−k}[k]+s_{i−k+1}[k]\). That is, because the final \(k−1\) symbols of \(s1\) overlap with the first \(k−1\) symbols of \(s2\), we simply tack on the k-th symbol of \(s2\) to \(s\), then iterate the process.
For example, the circular string assembled from the cycle “AC” → “CT” → “TA” → “AC” is simply (ACT). Note that this string only has length three because the 2-mers “wrap around” in the string.
If every k-mer in a collection of reads occurs as an edge in a de Bruijn graph cycle the same number of times as it appears in the reads, then we say that the cycle is “complete.”
Given: A list \(S_{k+1}\) of error-free DNA (k+1)-mers (\(k≤5\)) taken from the same strand of a circular chromosome (of length \(≤50\)).
Return: All circular strings assembled by complete cycles in the de Bruijn graph \(Bk\) of \(S_{k+1}\). The strings may be given in any order, but each one should begin with the first \((k+1)\)-mer provided in the input.
97.1 Sample Dataset
CAG
AGT
GTT
TTT
TTG
TGG
GGC
GCG
CGT
GTT
TTC
TCA
CAA
AAT
ATT
TTC
TCA97.2 Sample Output
CAGTTCAATTTGGCGTT
CAGTTCAATTGGCGTTT
CAGTTTCAATTGGCGTT
CAGTTTGGCGTTCAATT
CAGTTGGCGTTCAATTT
CAGTTGGCGTTTCAATT97.3 Solution
clas DeBruijnGraph:
clas Node:
def __init__(self, kmer):
self.kmer = kmer
self.neighbors = []
self.in_degree = 0
self.out_degree = 0
def __init__(self, reads):
self.graph = {}
self.start_kmer = reads[0]
for read in reads:
left_kmer, right_kmer = read[:-1], read[1:]
left_hash, right_hash = hash(left_kmer), hash(right_kmer)
left_node = self.graph.setdefault(left_hash, self.Node(left_kmer))
right_node = self.graph.setdefault(right_hash, self.Node(right_kmer))
left_node.neighbors.append(right_node)
left_node.out_degree += 1
right_node.in_degree += 1
def circular_string(self):
potential_starts = [node for node in self.graph.values() if node.out_degree > 1]
assert potential_starts, "No potential start nodes found!"
contigs = []
strings = set()
k = len(self.start_kmer) - 1
def dfs(node, sequence):
if node.out_degree > 1:
contigs.append(sequence + node.kmer[-1])
else:
dfs(node.neighbors[0], sequence + node.kmer[-1])
for start_node in potential_starts:
for neighbor in start_node.neighbors:
dfs(neighbor, start_node.kmer)
def find_circular_strings(current_sequence, accumulated_string, used_contigs):
if len(used_contigs) == len(contigs):
strings.add(accumulated_string)
else:
for idx in set(range(len(contigs))).difference(used_contigs):
if not current_sequence.endswith(contigs[idx][:k]):
continue
find_circular_strings(contigs[idx], accumulated_string + contigs[idx][:-k], used_contigs + (idx,))
for i, contig in enumerate(contigs):
if contig.startswith(self.start_kmer):
find_circular_strings(contig, contig[:-k], (i,))
break
return strings
# Sample input
sample_input = """
CAG
AGT
GTT
TTT
TTG
TGG
GGC
GCG
CGT
GTT
TTC
TCA
CAA
AAT
ATT
TTC
TCA
"""
reads = sample_input.strip().split("\n")
graph = DeBruijnGraph(reads)
print(*graph.circular_string(), sep='\n')97.4 Explanation
Graph Construction: The code constructs a De Bruijn graph using the provided k-mers (reads). Each k-mer’s prefix (all but the last character) and suffix (all but the first character) are treated as nodes in the graph. The graph edges represent transitions from one k-mer to another based on these prefixes and suffixes.
Node Structure: Each node in the graph stores its k-mer, its neighbors (other nodes it can connect to), and its in-degree and out-degree (how many edges enter and leave the node).
Graph Traversal: The code identifies nodes with more than one outgoing edge (
out_degree > 1) as potential starting points for generating circular sequences (possible cyclic paths in the graph).Depth-First Search (DFS): The code uses DFS to traverse from these potential starting nodes to build “contigs,” which are sequences representing possible paths through the graph.
Generating Circular Strings: After building contigs, the code recursively combines these contigs to generate complete circular strings that encompas all the original k-mers.
Output: The final set of circular strings that represent possible solutions is printed.
98 Finding a Motif with Modifications
Given a string s and a motif tt, an alignment of a substring of s against all of t is called a fitting alignment. Our aim is to find a substring \(s′\) of s that maximizes an alignment score with respect to \(t\).
Note that more than one such substring of \(s\) may exist, depending on the particular strings and alignment score used. One candidate for scoring function is the one derived from edit distance; In this problem, we will consider a slightly different alignment score, in which all matched symbols count as +1 and all mismatched symbols (including insertions and deletions) receive a cost of -1. Let’s call this scoring function the mismatch score.
Given: Two DNA strings \(s\) and \(t\), where \(s\) has length at most 10 kbp and \(t\) represents a motif of length at most 1 kbp.
Return: An optimal fitting alignment score with respect to the mismatch score defined above, followed by an optimal fitting alignment of a substring of s against tt. If multiple such alignments exist, then you may output any one.
98.1 Sample Dataset
>Rosalind_54
GCAAACCATAAGCCCTACGTGCCGCCTGTTTAAACTCGCGAACTGAATCTTCTGCTTCACGGTGAAAGTACCACAATGGTATCACACCCCAAGGAAAC
>Rosalind_46
GCCGTCAGGCTGGTGTCCG98.2 Sample Output
5
ACCATAAGCCCTACGTG-CCG
GCCGTCAGGC-TG-GTGTCCG98.3 Solution
from typing import List, Tuple
GAP_PENALTY = 1
MATCH_SCORE = 1
MISMATCH_PENALTY = 1
def parse_fasta(data: str) -> List[str]:
"""Parse FASTA format data into a list of sequences."""
sequences = []
current_seq = []
for line in data.strip().split('\n'):
if line.startswith('>'):
if current_seq:
sequences.append(''.join(current_seq))
current_seq = []
else:
current_seq.append(line.strip())
if current_seq:
sequences.append(''.join(current_seq))
return sequences
def initialize_dp_matrix(m: int, n: int) -> List[List[int]]:
"""Initialize the dynamic programming matrix."""
return [[0 for _ in range(n + 1)] for _ in range(m + 1)]
def fill_dp_matrix(c: str, d: str, dp: List[List[int]]) -> None:
"""Fill the dynamic programming matrix."""
for i in range(len(c) + 1):
for j in range(1, len(d) + 1):
ans = float('-inf')
if i > 0:
ans = max(ans, dp[i - 1][j] - GAP_PENALTY)
if j > 0:
ans = max(ans, dp[i][j - 1] - GAP_PENALTY)
if i > 0 and j > 0:
if c[i - 1] == d[j - 1]:
ans = max(ans, dp[i - 1][j - 1] + MATCH_SCORE)
else:
ans = max(ans, dp[i - 1][j - 1] - MISMATCH_PENALTY)
dp[i][j] = ans
def find_best_score(dp: List[List[int]], m: int, n: int) -> Tuple[int, int, int]:
"""Find the best score and its position in the DP matrix."""
score = float('-inf')
bi, bj = -1, -1
for i in range(m + 1):
if score < dp[i][n]:
score = dp[i][n]
bi, bj = i, n
return int(score), bi, bj
def backtrack(c: str, d: str, dp: List[List[int]], bi: int, bj: int) -> Tuple[str, str]:
"""Backtrack to find the aligned sequences."""
s1, s2 = [], []
while bj > 0:
if bi > 0 and dp[bi - 1][bj] - GAP_PENALTY == dp[bi][bj]:
s1.append(c[bi - 1])
s2.append('-')
bi -= 1
elif bj > 0 and dp[bi][bj - 1] - GAP_PENALTY == dp[bi][bj]:
s1.append('-')
s2.append(d[bj - 1])
bj -= 1
else:
s1.append(c[bi - 1])
s2.append(d[bj - 1])
bi -= 1
bj -= 1
return ''.join(s1[::-1]), ''.join(s2[::-1])
sample_input = """
>Rosalind_54
GCAAACCATAAGCCCTACGTGCCGCCTGTTTAAACTCGCGAACTGAATCTTCTGCTTCACGGTGAAAGTACCACAATGGTATCACACCCCAAGGAAAC
>Rosalind_46
GCCGTCAGGCTGGTGTCCG
"""
sequences = parse_fasta(sample_input)
c, d = sequences
dp = initialize_dp_matrix(len(c), len(d))
fill_dp_matrix(c, d, dp)
score, bi, bj = find_best_score(dp, len(c), len(d))
aligned_c, aligned_d = backtrack(c, d, dp, bi, bj)
print(score)
print(aligned_c)
print(aligned_d)This code performs a sequence alignment between two DNA sequences using a dynamic programming approach. Sequence alignment is a method used in bioinformatics to compare two sequences and determine the best match between them, accounting for matches, mismatches, and gaps.
98.4 How the Code Works
Constants:
GAP_PENALTY: Penalty for introducing a gap (insertion/deletion) in the sequence alignment.MATCH_SCORE: Score for matching characters between the two sequences.MISMATCH_PENALTY: Penalty for mismatched characters.
Functions:
parse_fasta(data: str) -> List[str]:- Purpose: Converts a FASTA format string into a list of sequences.
- How It Works: The function reads the input data, ignoring lines starting with ‘>’ (which are headers), and combines the remaining lines into sequences.
initialize_dp_matrix(m: int, n: int) -> List[List[int]]:- Purpose: Initializes a matrix for dynamic programming (DP) with dimensions
(m+1) x (n+1), wheremandnare the lengths of the two sequences. - How It Works: Creates a 2D list filled with zeros.
- Purpose: Initializes a matrix for dynamic programming (DP) with dimensions
fill_dp_matrix(c: str, d: str, dp: List[List[int]]) -> None:- Purpose: Fills the DP matrix with scores based on the alignment of sequences
candd. - How It Works:
- Iterates over all possible alignments of
candd. - For each position, it calculates the best score considering three possible moves: match/mismatch, insertion, and deletion.
- The score is updated based on whether characters match or mismatch, and whether gaps are introduced.
- Iterates over all possible alignments of
- Purpose: Fills the DP matrix with scores based on the alignment of sequences
find_best_score(dp: List[List[int]], m: int, n: int) -> Tuple[int, int, int]:- Purpose: Finds the best alignment score and its position in the DP matrix.
- How It Works:
- Scans the last column of the DP matrix to find the highest score and its position. This represents the optimal alignment score.
backtrack(c: str, d: str, dp: List[List[int]], bi: int, bj: int) -> Tuple[str, str]:- Purpose: Traces back through the DP matrix to construct the aligned sequences based on the best alignment score.
- How It Works:
- Starting from the best score position, it determines the path that led to this score, reconstructing the aligned sequences with gaps (
-) where necessary.
- Starting from the best score position, it determines the path that led to this score, reconstructing the aligned sequences with gaps (
Execution:
- Sample Input: The FASTA input contains two sequences labeled
Rosalind_54andRosalind_46. - Processing:
- The sequences are extracted using
parse_fasta. - A DP matrix is initialized using
initialize_dp_matrix. - The matrix is filled with alignment scores using
fill_dp_matrix. - The best alignment score and position are found with
find_best_score. - The best alignment itself is reconstructed using
backtrack.
- The sequences are extracted using
- Output:
- The alignment score and the two aligned sequences are printed.
- Sample Input: The FASTA input contains two sequences labeled
99 Isolating Symbols in Alignments
Say that we have two strings s and t of respective lengths \(m\) and \(n\) and an alignment score. Let’s define a matrix MM corresponding to s and t by setting \(M_{j,k}\) equal to the maximum score of any alignment that aligns \(s[j]\) with \(t[k]\). So each entry in \(M\) can be equal to at most the maximum score of any alignment of s and \(t\).
Given: Two DNA strings \(s\) and \(t\) in FASTA format, each having length at most 1000 bp.
Return: The maximum alignment score of a global alignment of \(s\) and \(t\), followed by the sum of all elements of the matrix \(M\) corresponding to \(s\) and \(t\) that was defined above. Apply the mismatch score introduced in “Finding a Motif with Modifications”.
99.1 Sample Dataset
>Rosalind_35
ATAGATA
>Rosalind_5
ACAGGTA99.2 Sample Output
3
-13999.3 Solution
from typing import List, Tuple
def parse_fasta(data: str) -> List[str]:
"""Parse FASTA format data into a list of sequences."""
sequences = []
current_seq = []
for line in data.strip().split('\n'):
if line.startswith('>'):
if current_seq:
sequences.append(''.join(current_seq))
current_seq = []
else:
current_seq.append(line.strip())
if current_seq:
sequences.append(''.join(current_seq))
return sequences
def initialize_score_matrix(rows: int, cols: int) -> List[List[int]]:
"""Initialize the score matrix with gap penalties."""
S = [[0 for _ in range(cols)] for _ in range(rows)]
for i in range(1, rows):
S[i][0] = -i
for j in range(1, cols):
S[0][j] = -j
return S
def calculate_cell_score(S: List[List[int]], i: int, j: int, s: str, t: str) -> int:
"""Calculate the score for a cell in the alignment matrix."""
match_score = 1 if s[i-1] == t[j-1] else -1
return max(
S[i-1][j-1] + match_score,
S[i-1][j] - 1,
S[i][j-1] - 1
)
def global_alignment(s: str, t: str) -> List[List[int]]:
"""Perform global alignment and return the score matrix."""
rows, cols = len(s) + 1, len(t) + 1
S = initialize_score_matrix(rows, cols)
for i in range(1, rows):
for j in range(1, cols):
S[i][j] = calculate_cell_score(S, i, j, s, t)
return S
def align_to_symbols(s: str, t: str) -> Tuple[int, int]:
"""Compute the maximum alignment score and sum of all alignment scores."""
prefix_matrix = global_alignment(s, t)
suffix_matrix = global_alignment(s[::-1], t[::-1])
total = 0
best = -(len(s) + len(t))
for i in range(len(s)):
for j in range(len(t)):
match_score = 1 if s[i] == t[j] else -1
score = prefix_matrix[i][j] + match_score + suffix_matrix[len(s)-1-i][len(t)-1-j]
total += score
best = max(best, score)
return best, total
sample_input = """
>Rosalind_35
ATAGATA
>Rosalind_5
ACAGGTA
"""
sequences = parse_fasta(sample_input)
if len(sequences) != 2:
raise ValueError("Expected exactly two sequences in the input.")
s, t = sequences
best_score, total_score = align_to_symbols(s, t)
print(f"{best_score}")
print(f"{total_score}")99.4 Code Breakdown
Parsing FASTA Format (
parse_fastafunction):- Purpose: Extracts DNA sequences from a FASTA formatted string.
- How It Works:
- Input: A string with lines that include sequences starting with
>(sequence headers) and followed by sequence data. - Process:
- It reads the input line by line.
- When it encounters a line starting with
>, it recognizes it as a header and finishes the current sequence. - It collects sequence data lines and joins them into a single sequence string.
- It returns a list of sequences.
- Input: A string with lines that include sequences starting with
- Output: A list of DNA sequences.
Initializing the Score Matrix (
initialize_score_matrixfunction):- Purpose: Set up a matrix to track alignment scores between two sequences, initializing with gap penalties.
- How It Works:
- Input: Number of rows (sequence length + 1) and columns (sequence length + 1).
- Process:
- Creates a matrix of zeros.
- Fills the first row and column with penalties for gaps (negative values), representing the cost of inserting gaps.
- Output: A score matrix with initialized gap penalties.
Calculating Cell Scores (
calculate_cell_scorefunction):- Purpose: Determine the alignment score for a specific cell in the matrix.
- How It Works:
- Input: Current cell indices, the score matrix, and the sequences being aligned.
- Process:
- Calculates the score for matching or mismatching characters, and the penalty for gaps.
- Takes the maximum score among possible scenarios: match/mismatch, gap in one sequence, or gap in the other sequence.
- Output: The best score for the current cell.
Global Alignment (
global_alignmentfunction):- Purpose: Create and fill a score matrix for global alignment of two sequences.
- How It Works:
- Input: Two sequences.
- Process:
- Initializes the score matrix with gap penalties.
- Fills the matrix using
calculate_cell_scorefor each cell.
- Output: A filled score matrix representing global alignment scores.
Aligning to Symbols (
align_to_symbolsfunction):- Purpose: Calculate the best alignment score and total score sum by considering both the original and reversed sequences.
- How It Works:
- Input: Two sequences.
- Process:
- Computes the alignment score matrices for both the original sequences and their reversed versions.
- Calculates scores by combining the prefix (forward alignment) and suffix (reverse alignment) matrices.
- Finds the best score and sums all scores from the matrix.
- Output: The highest alignment score and the total sum of all scores.
100 Finding All Similar Motifs
Given: A positive integer \(k\) (\(k≤50\)), a DNA string s of length at most 5 kbp representing a motif, and a DNA string t of length at most 50 kbp representing a genome.
Return: All substrings \(t′\) of \(t\) such that the edit distance \(dE(s,t′)\) is les than or equal to \(k\). Each substring should be encoded by a pair containing its location in \(t\) followed by its length.
100.1 Sample Dataset
2
ACGTAG
ACGGATCGGCATCGT100.2 Sample Output
1 4
1 5
1 6100.3 Solution
import sys
import re
import multiprocessing as mp
def get_seeds(x, seq, k):
seed_size = len(x) // (k + 1)
for s1 in range(0, len(x) - seed_size + 1, seed_size):
px = (s1, s1 + seed_size)
seed = x[px[0] : px[1]]
for m in re.finditer(rf"(?=({seed}))", seq):
ps = (m.span()[0], m.span()[0] + seed_size)
yield (px, ps)
def process_seed(args):
def extend_fwd(i, j, score):
if (i, j, score) not in seen:
seen.update([(i, j, score)])
if score <= k:
if i == len(x) - 1:
yield i, j, score
if i + 1 < len(x):
yield from extend_fwd(i + 1, j, score + 1)
if j + 1 < len(seq):
yield from extend_fwd(i, j + 1, score + 1)
if i + 1 < len(x) and j + 1 < len(seq):
yield from extend_fwd(
i + 1, j + 1, score + int(x[i + 1] != seq[j + 1])
)
def extend_rev(i, j, score):
if (i, j, score) not in seen:
seen.update([(i, j, score)])
if score <= k:
if i == 0:
yield i, j, score
if i - 1 >= 0:
yield from extend_rev(i - 1, j, score + 1)
if j - 1 >= 0:
yield from extend_rev(i, j - 1, score + 1)
if i - 1 >= 0 and j - 1 >= 0:
yield from extend_rev(
i - 1, j - 1, score + int(x[i - 1] != seq[j - 1])
)
print(".", end="", file=sys.stderr)
sys.stderr.flush()
sys.setrecursionlimit(10000)
seed, k, x, seq = args
xcoord, seqcoord = seed
res = set()
seen = set()
fwds = list(extend_fwd(xcoord[1] - 1, seqcoord[1] - 1, 0))
if not fwds:
return set()
seen = set()
revs = list(extend_rev(xcoord[0], seqcoord[0], 0))
if not revs:
return set()
for i0, j0, s0 in revs:
for i1, j1, s1 in fwds:
if s0 + s1 <= k:
res.add((j0 + 1, j1 - j0 + 1))
return res
# Sample input
sample_input = """
1
ACGTAG
GGACGATAGGTAAAGTAGTAGCGACGTAGG
"""
k, x, seq = sample_input.strip().split("\n")
k = int(k)
seeds = list(get_seeds(x, seq, k))
print(f"found {len(seeds)} seeds", file=sys.stderr)
pool = mp.Pool(mp.cpu_count())
args = ([seed, k, x, seq] for seed in seeds)
res = pool.map(process_seed, args)
res = set().union(*res)
# 결과 출력
for start, length in sorted(list(res)):
print(f"{start} {length}")해당 문제는 계산량이 많아 파이썬을 사용하는 것이 적절하지 않습니다. 그래도 위 코드를 download dataset 에 적용해 실행한 결과 1 분 45 초 정도가 소요되어 통과할 수 있었습니다.(사용한 CPU: 13th Gen Intel i9-13900F (32) @ 5.3GHz)
This Python code is designed to find approximate matches of a short DNA sequence x within a longer DNA sequence seq. It does this by breaking down the problem into smaller “seed” sequences and then extending those seeds to find matches, even if there are a few mismatches allowed (controlled by the parameter k).
100.4 Overview of the Code
- Seeding (
get_seedsfunction):- The
get_seedsfunction divides the sequencexinto smaller overlapping segments called “seeds.” - For each seed, it searches within the longer sequence
seqto find exact matches of the seed. - It returns the positions in both
xandseqwhere these seeds match.
- The
- Extending Seeds (
process_seedfunction):- This function takes a seed and tries to extend it in both directions (forward and backward) to see if a longer match can be found between
xandseq, even with up tokmismatches. - Two helper functions,
extend_fwdandextend_rev, recursively extend the seed by comparing characters inxandseqwhile keeping track of mismatches. - The results are stored as starting positions and lengths of the matching segments.
- This function takes a seed and tries to extend it in both directions (forward and backward) to see if a longer match can be found between
- Parallel Processing:
- The script uses multiprocessing to speed up the search by running the seed extension proces in parallel acros multiple CPU cores.
- Each seed is processed independently, and the results are combined.
- Result Compilation:
- The final matching segments are collected, and the unique results are sorted and printed as the start position and length of each matching segment in
seq.
- The final matching segments are collected, and the unique results are sorted and printed as the start position and length of each matching segment in
- The code searches for parts of a short DNA sequence (
x) within a longer DNA sequence (seq), allowing for a small number of mismatches (k). - It does this by first finding small exact matches (seeds) and then extending these matches to find longer sequences with few mismatches.
- The proces is parallelized to improve performance, especially when dealing with large DNA sequences.
101 Overlap Alignment
An overlap alignment between two strings \(s\) and \(t\) is a local alignment of a suffix of \(s\) with a prefix of \(t\). An optimal overlap alignment will therefore maximize an alignment score over all such substrings of \(s\) and \(t\).
The term “overlap alignment” has also been used to describe what Rosalind defines as a semiglobal alignment. See “Semiglobal Alignment” for details.
Given: Two DNA strings \(s\) and \(t\) in FASTA format, each having length at most 10 kbp.
Return: The score of an optimal overlap alignment of s and tt, followed by an alignment of a suffix \(s′\) of s and a prefix \(t′\) of t achieving this optimal score. Use an alignment score in which matching symbols count +1, substitutions count -2, and there is a linear gap penalty of 2. If multiple optimal alignments exist, then you may return any one.
101.1 Sample Dataset
>Rosalind_54
CTAAGGGATTCCGGTAATTAGACAG
>Rosalind_45
ATAGACCATATGTCAGTGACTGTGTAA101.2 Sample Output
1
ATTAGAC-AG
AT-AGACCAT101.3 Solution
import numpy as np
def oap(s1, s2, penalty=-2):
score = np.empty((len(s2) + 1, len(s1) + 1), dtype=int)
ptr = np.empty((len(s2) + 1, len(s1) + 1), dtype=int)
for j in range(len(s2) + 1):
score[j][0] = j * penalty
ptr[j][0] = 1
for i in range(len(s1) + 1):
score[0][i] = 0
ptr[0][i] = 2
score[0][0] = 0
for j in range(len(s2)):
for i in range(len(s1)):
opt = [
score[j][i] + (1 if s1[i] == s2[j] else penalty),
score[j][i + 1] + penalty,
score[j + 1][i] + penalty,
]
best = max(opt)
score[j + 1][i + 1] = best
ptr[j + 1][i + 1] = opt.index(best)
sc = [score[j][len(s1)] for j in range(len(s2) + 1)]
max_score = max(sc)
j = [j for j, s in enumerate(sc) if s == max_score][-1]
i = len(s1)
a1, a2 = "", ""
while i > 0 and j > 0:
if ptr[j][i] == 0:
a1 += s1[i - 1]
a2 += s2[j - 1]
j, i = j - 1, i - 1
elif ptr[j][i] == 1:
a1 += "-"
a2 += s2[j - 1]
j = j - 1
elif ptr[j][i] == 2:
a1 += s1[i - 1]
a2 += "-"
i = i - 1
return max_score, a1[::-1], a2[::-1]
def read_fasta(fasta_string):
"""
Parses a FASTA formatted string and returns a list of sequences.
"""
sequences = []
current_sequence = []
for line in fasta_string.strip().split('\n'):
if line.startswith('>'):
if current_sequence:
sequences.append(''.join(current_sequence))
current_sequence = []
else:
current_sequence.append(line.strip())
if current_sequence:
sequences.append(''.join(current_sequence))
return sequences
sample_input = """
>Rosalind_54
CTAAGGGATTCCGGTAATTAGACAG
>Rosalind_45
ATAGACCATATGTCAGTGACTGTGTAA
"""
s1, s2 = read_fasta(sample_input)
print(*oap(s1, s2, -2), sep="\n")This code implements a semi-global sequence alignment algorithm, also known as overlap alignment. Here’s a brief explanation of how it works:
- Initialization:
- Creates two matrices: ‘score’ for alignment scores and ‘ptr’ for backtracking.
- Initializes the first row and column of these matrices.
- Filling the matrices:
- Iterates through both sequences, filling the ‘score’ and ‘ptr’ matrices.
- For each cell, calculates three possible scores: match/mismatch, gap in s1, gap in s2.
- Chooses the maximum score and stores it along with a pointer to its origin.
- Finding the best alignment:
- Finds the maximum score in the last column of the ‘score’ matrix.
- This allows for free end gaps in s2 (overlap alignment).
- Traceback:
- Starts from the position of the maximum score.
- Follows the pointers back to construct the aligned sequences.
- Adds gaps (‘-’) where necessary.
- Result:
- Returns the maximum alignment score and the two aligned sequences.
The ‘read_fasta’ function parses a FASTA-formatted string into sequences.
Finally, it applies this alignment algorithm to two sequences from the sample input and prints the results.
This algorithm is particularly useful for finding the best overlap between two sequences, allowing for free end gaps in one of the sequences.
102 Quartet Distance
In “Counting Quartets”, we found an expression for \(q(T)\), the number of quartets that can be inferred from an unrooted binary tree containing \(n\) taxa.
If \(T1\) and \(T2\) are both unrooted binary trees on the same \(n\) taxa, then we now let \(q(T1,T2)\) denote the number of inferred quartets that are common to both trees. The quartet distance between \(T1\) and \(T2\), \(dq(T1,T2)\) is the number of quartets that are only inferred from one of the trees. More precisely, \(dq(T1,T2)=q(T1)+q(T2)−2q(T1,T2)\).
Given: A list containing \(n\) taxa (\(n≤2000\)) and two unrooted binary trees \(T1\) and \(T2\) on the given taxa. Both \(T1\) and \(T2\) are given in Newick format.
Return: The quartet distance \(dq(T1,T2)\).
102.1 Sample Dataset
A B C D E
(A,C,((B,D),E));
(C,(B,D),(A,E));102.2 Sample Output
4102.3 Solution
import re
import math
def quartet_distance(taxa, t1, t2):
def parse_newick(taxa, tree_str):
tree = {}
parent = {}
node_names = {}
new_node_id = 0
root = new_node_id
tree[root] = []
current_node = root
for match in re.finditer(r"\(|\)|,|;|([^\(\),;]+)", tree_str):
token = match.group()
if token == '(':
new_node_id += 1
tree[current_node].append(new_node_id)
parent[new_node_id] = current_node
current_node = new_node_id
tree[current_node] = []
elif token == ',':
new_node_id += 1
tree[parent[current_node]].append(new_node_id)
parent[new_node_id] = parent[current_node]
current_node = new_node_id
tree[current_node] = []
elif token == ')':
current_node = parent[current_node]
elif token == ';':
break
else:
node_names[current_node] = token
return tree, parent, node_names
def get_children(tree, parent, edge_id):
if edge_id > 0:
return tree[edge_id]
else:
parent_id = parent[-edge_id]
if parent_id == 0:
return [e for e in tree[parent_id] if e != -edge_id]
else:
return [-parent_id] + [e for e in tree[parent_id] if e != -edge_id]
def compute_shared_leaves(i, j):
if shared_leaves[i][j] is None:
if not children1[i] and not children2[j]: # Both are leaves
shared_leaves[i][j] = int(leaves1[i] == leaves2[j])
elif not children1[i]: # i is a leaf
j1, j2 = children2[j]
shared_leaves[i][j] = compute_shared_leaves(i, j1) + compute_shared_leaves(i, j2)
elif not children2[j]: # j is a leaf
i1, i2 = children1[i]
shared_leaves[i][j] = compute_shared_leaves(i1, j) + compute_shared_leaves(i2, j)
else: # Both are internal nodes
i1, i2 = children1[i]
j1, j2 = children2[j]
shared_leaves[i][j] = (
compute_shared_leaves(i1, j1) + compute_shared_leaves(i1, j2) +
compute_shared_leaves(i2, j1) + compute_shared_leaves(i2, j2)
)
return shared_leaves[i][j]
def calculate_quartet_distances():
for i in all_edges1:
for j in all_edges2:
compute_shared_leaves(i, j)
total_distance = 0
for c1 in internal_edges1:
for c2 in internal_edges2:
a1, b1 = children1[-c1]
a2, b2 = children2[-c2]
quartet_value = (
shared_leaves[a1][a2] * shared_leaves[b1][b2] +
shared_leaves[a1][b2] * shared_leaves[b1][a2]
)
total_distance += quartet_value * (shared_leaves[c1][c2] * (shared_leaves[c1][c2] - 1) / 2)
return total_distance
# Parse Newick trees
tree1, parent1, leaves1 = parse_newick(taxa, t1)
tree2, parent2, leaves2 = parse_newick(taxa, t2)
# Number of taxa
n = len(taxa)
# Get children of each edge
children1 = [None] * (4 * n - 5)
children2 = [None] * (4 * n - 5)
for i in range(1, 2 * n - 2):
children1[i] = get_children(tree1, parent1, i)
for i in range(3 - 2 * n, 0):
children1[i] = get_children(tree1, parent1, i)
for j in range(1, 2 * n - 2):
children2[j] = get_children(tree2, parent2, j)
for j in range(3 - 2 * n, 0):
children2[j] = get_children(tree2, parent2, j)
# Initialize shared leaves matrix
shared_leaves = [[None] * (4 * n - 5) for _ in range(4 * n - 5)]
# List of all edges and internal edges
all_edges1 = list(range(1, 2 * n - 2)) + [edge for edge in range(3 - 2 * n, 0) if leaves1.get(-edge) is None]
all_edges2 = list(range(1, 2 * n - 2)) + [edge for edge in range(3 - 2 * n, 0) if leaves2.get(-edge) is None]
internal_edges1 = [edge for edge in all_edges1 if leaves1.get(edge) is None]
internal_edges2 = [edge for edge in all_edges2 if leaves2.get(edge) is None]
# Calculate quartet distances
total_quartets = calculate_quartet_distances()
# Calculate and return the quartet distance
max_possible_quartets = 2 * math.comb(n, 4)
return max_possible_quartets - total_quartets
# Sample input
sample_input = """
A B C D E
(A,C,((B,D),E));
(C,(B,D),(A,E));
""".strip().split("\n")
taxa = sample_input[0].split()
nwck1 = sample_input[1]
nwck2 = sample_input[2]
print(quartet_distance(taxa, nwck1, nwck2))102.4 Explanation of How It Works
- Newick Parsing (
parse_newick):- The function
parse_newickparses a Newick-formatted tree string and constructs a representation of the tree using a dictionarytree, which maps node IDs to their children. It also maintains aparentdictionary to track parent-child relationships and anamedictionary to map node IDs to taxa names. - This parsed tree allows us to later traverse and compare the structures of the two trees.
- The function
- Children Function (
get_children):- The
get_childrenfunction retrieves the children of a given edge in the tree. If the edge is positive, it directly retrieves children from the tree structure. If the edge is negative, it handles the reversed direction by excluding the edge itself from the parent’s list.
- The
- Shared Leaves Calculation (
compute_shared_leaves):- The
compute_shared_leavesfunction computes the number of shared leaves between two subtrees (one from each tree) by recursively exploring their child nodes. It caches results to avoid redundant calculations, significantly optimizing performance.
- The
- Quartet Calculation (
calculate_quartet_distances):- The
calculate_quartet_distancesfunction iterates over all pairs of internal edges from the two trees and calculates the quartet distances. A quartet distance measures the difference in tree structure by comparing the shared leaves for each quartet configuration. - This function sums up these quartet values, representing the differences between the two trees.
- The
- Quartet Distance Calculation:
- Finally, the
quartet_distancefunction subtracts the calculated quartet differences from the total possible quartets for the number of taxa (given bymath.comb(n, 4)). This provides the quartet distance, a measure of how dissimilar the two trees are in terms of their quartets.
- Finally, the
103 Semiglobal Alignment
A semiglobal alignment of strings s and t is an alignment in which any gaps appearing as prefixes or suffixes of \(s\) and \(t\) do not contribute to the alignment score.
Semiglobal alignment has sometimes also been called “overlap alignment”. Rosalind defines overlap alignment differently (see “Overlap Alignment”).
Given: Two DNA strings s and t in FASTA format, each having length at most 10 kbp.
Return: The maximum semiglobal alignment score of \(s\) and \(t\), followed by an alignment of \(s\) and \(t\) achieving this maximum score. Use an alignment score in which matching symbols count +1, substitutions count -1, and there is a linear gap penalty of 1. If multiple optimal alignments exist, then you may return any one.
103.1 Sample Dataset
>Rosalind_79
CAGCACTTGGATTCTCGG
>Rosalind_98
CAGCGTGG103.2 Sample Output
4
CAGCA-CTTGGATTCTCGG
---CAGCGTGG--------103.3 Solution
def semiglobal_alignment(seq1, seq2):
seq1 = "-" + seq1
seq2 = "-" + seq2
score_matrix = [[0 for j in range(len(seq2))] for i in range(len(seq1))]
direction_matrix = [[None for j in range(len(seq2))] for i in range(len(seq1))]
for i in range(1, len(seq1)):
for j in range(1, len(seq2)):
match_score = score_matrix[i - 1][j - 1] + (1 if seq1[i] == seq2[j] else -1)
delete_score = score_matrix[i - 1][j] - 1
insert_score = score_matrix[i][j - 1] - 1
score_matrix[i][j] = max(match_score, delete_score, insert_score)
if score_matrix[i][j] == match_score:
direction_matrix[i][j] = "diagonal"
elif score_matrix[i][j] == delete_score:
direction_matrix[i][j] = "up"
else:
direction_matrix[i][j] = "left"
last_row_max = max(range(len(seq2)), key=lambda x: score_matrix[len(seq1) - 1][x])
last_col_max = max(range(len(seq1)), key=lambda x: score_matrix[x][len(seq2) - 1])
if score_matrix[len(seq1) - 1][last_row_max] >= score_matrix[last_col_max][len(seq2) - 1]:
i = len(seq1) - 1
j = last_row_max
else:
i = last_col_max
j = len(seq2) - 1
max_score = score_matrix[i][j]
insert_gap = lambda word, i: word[:i] + '-' + word[i:]
# Initialize the aligned sequences as the input sequences.
aligned_seq1, aligned_seq2 = seq1[1:], seq2[1:]
for _ in range(len(seq1) - 1 - i):
aligned_seq2 += '-'
for _ in range(len(seq2) - 1 - j):
aligned_seq1 += '-'
while i * j != 0:
if direction_matrix[i][j] == "up":
i -= 1
aligned_seq2 = insert_gap(aligned_seq2, j)
elif direction_matrix[i][j] == "left":
j -= 1
aligned_seq1 = insert_gap(aligned_seq1, i)
else:
i -= 1
j -= 1
for _ in range(i):
aligned_seq2 = insert_gap(aligned_seq2, 0)
for _ in range(j):
aligned_seq1 = insert_gap(aligned_seq1, 0)
return max_score, aligned_seq1, aligned_seq2
def parse_fasta(fasta_string):
sequences = {}
current_label = None
for line in fasta_string.strip().split('\n'):
if line.startswith('>'):
current_label = line[1:].strip()
sequences[current_label] = ''
else:
sequences[current_label] += line.strip()
return list(sequences.values())
sample_input = """
>Rosalind_79
CAGCACTTGGATTCTCGG
>Rosalind_98
CAGCGTGG
"""
sequence_A, sequence_B = parse_fasta(sample_input)
final_score, aligned_sequence_A, aligned_sequence_B = semiglobal_alignment(sequence_A, sequence_B)
print(final_score)
print(aligned_sequence_A)
print(aligned_sequence_B)The
semiglobal_alignmentfunction implements a semi-global alignment algorithm for two sequences:- It adds a gap character “-” at the beginning of both sequences.
- It creates two matrices:
score_matrixfor alignment scores anddirection_matrixfor backtracking.
The function then fills these matrices:
- It calculates scores for matches (1), mismatches (-1), and gaps (-1).
- It chooses the maximum score among match, deletion, and insertion for each cell.
- It records the direction (diagonal, up, or left) in the
direction_matrix.
After filling the matrices, it finds the best alignment end point:
- It checks the maximum score in the last row and last column.
- It chooses the higher of these two as the ending point of the alignment.
The function then performs a traceback to construct the aligned sequences:
- It starts from the best end point and follows the directions in
direction_matrix. - It adds gaps to the sequences as needed during the traceback.
- It starts from the best end point and follows the directions in
Finally, it returns the maximum score and the two aligned sequences.
The
parse_fastafunction reads a FASTA-formatted string:- It separates the sequences and their labels.
- It returns a list of sequences without the labels.
The main part of the code:
- Defines a sample input in FASTA format.
- Parses the input using
parse_fasta. - Calls
semiglobal_alignmentwith the parsed sequences. - Prints the final score and the aligned sequences.
104 Local Alignment with Affine Gap Penalty
Given: Two protein strings \(s\) and \(t\) in FASTA format (each having length at most 10,000 aa).
Return: The maximum local alignment score of \(s\) and \(t\), followed by substrings \(r\) and \(u\) of \(s\) and \(t\), respectively, that correspond to the optimal local alignment of \(s\) and \(t\). Use:
- The BLOSUM62 scoring matrix.
- Gap opening penalty equal to 11.
- Gap extension penalty equal to 1.
If multiple solutions exist, then you may output any one.
104.1 Sample Dataset
>Rosalind_8
PLEASANTLY
>Rosalind_18
MEANLY104.2 Sample Output
12
LEAS
MEAN104.3 Solution
BLOSUM62 = {
('W', 'F'): 1, ('L', 'R'): -2, ('S', 'P'): -1, ('V', 'T'): 0,
('Q', 'Q'): 5, ('N', 'A'): -2, ('Z', 'Y'): -2, ('W', 'R'): -3,
('Q', 'A'): -1, ('S', 'D'): 0, ('H', 'H'): 8, ('S', 'H'): -1,
('H', 'D'): -1, ('L', 'N'): -3, ('W', 'A'): -3, ('Y', 'M'): -1,
('G', 'R'): -2, ('Y', 'I'): -1, ('Y', 'E'): -2, ('B', 'Y'): -3,
('Y', 'A'): -2, ('V', 'D'): -3, ('B', 'S'): 0, ('Y', 'Y'): 7,
('G', 'N'): 0, ('E', 'C'): -4, ('Y', 'Q'): -1, ('Z', 'Z'): 4,
('V', 'A'): 0, ('C', 'C'): 9, ('M', 'R'): -1, ('V', 'E'): -2,
('T', 'N'): 0, ('P', 'P'): 7, ('V', 'I'): 3, ('V', 'S'): -2,
('Z', 'P'): -1, ('V', 'M'): 1, ('T', 'F'): -2, ('V', 'Q'): -2,
('K', 'K'): 5, ('P', 'D'): -1, ('I', 'H'): -3, ('I', 'D'): -3,
('T', 'R'): -1, ('P', 'L'): -3, ('K', 'G'): -2, ('M', 'N'): -2,
('P', 'H'): -2, ('F', 'Q'): -3, ('Z', 'G'): -2, ('X', 'L'): -1,
('T', 'M'): -1, ('Z', 'C'): -3, ('X', 'H'): -1, ('D', 'R'): -2,
('B', 'W'): -4, ('X', 'D'): -1, ('Z', 'K'): 1, ('F', 'A'): -2,
('Z', 'W'): -3, ('F', 'E'): -3, ('D', 'N'): 1, ('B', 'K'): 0,
('X', 'X'): -1, ('F', 'I'): 0, ('B', 'G'): -1, ('X', 'T'): 0,
('F', 'M'): 0, ('B', 'C'): -3, ('Z', 'I'): -3, ('Z', 'V'): -2,
('S', 'S'): 4, ('L', 'Q'): -2, ('W', 'E'): -3, ('Q', 'R'): 1,
('N', 'N'): 6, ('W', 'M'): -1, ('Q', 'C'): -3, ('W', 'I'): -3,
('S', 'C'): -1, ('L', 'A'): -1, ('S', 'G'): 0, ('L', 'E'): -3,
('W', 'Q'): -2, ('H', 'G'): -2, ('S', 'K'): 0, ('Q', 'N'): 0,
('N', 'R'): 0, ('H', 'C'): -3, ('Y', 'N'): -2, ('G', 'Q'): -2,
('Y', 'F'): 3, ('C', 'A'): 0, ('V', 'L'): 1, ('G', 'E'): -2,
('G', 'A'): 0, ('K', 'R'): 2, ('E', 'D'): 2, ('Y', 'R'): -2,
('M', 'Q'): 0, ('T', 'I'): -1, ('C', 'D'): -3, ('V', 'F'): -1,
('T', 'A'): 0, ('T', 'P'): -1, ('B', 'P'): -2, ('T', 'E'): -1,
('V', 'N'): -3, ('P', 'G'): -2, ('M', 'A'): -1, ('K', 'H'): -1,
('V', 'R'): -3, ('P', 'C'): -3, ('M', 'E'): -2, ('K', 'L'): -2,
('V', 'V'): 4, ('M', 'I'): 1, ('T', 'Q'): -1, ('I', 'G'): -4,
('P', 'K'): -1, ('M', 'M'): 5, ('K', 'D'): -1, ('I', 'C'): -1,
('Z', 'D'): 1, ('F', 'R'): -3, ('X', 'K'): -1, ('Q', 'D'): 0,
('X', 'G'): -1, ('Z', 'L'): -3, ('X', 'C'): -2, ('Z', 'H'): 0,
('B', 'L'): -4, ('B', 'H'): 0, ('F', 'F'): 6, ('X', 'W'): -2,
('B', 'D'): 4, ('D', 'A'): -2, ('S', 'L'): -2, ('X', 'S'): 0,
('F', 'N'): -3, ('S', 'R'): -1, ('W', 'D'): -4, ('V', 'Y'): -1,
('W', 'L'): -2, ('H', 'R'): 0, ('W', 'H'): -2, ('H', 'N'): 1,
('W', 'T'): -2, ('T', 'T'): 5, ('S', 'F'): -2, ('W', 'P'): -4,
('L', 'D'): -4, ('B', 'I'): -3, ('L', 'H'): -3, ('S', 'N'): 1,
('B', 'T'): -1, ('L', 'L'): 4, ('Y', 'K'): -2, ('E', 'Q'): 2,
('Y', 'G'): -3, ('Z', 'S'): 0, ('Y', 'C'): -2, ('G', 'D'): -1,
('B', 'V'): -3, ('E', 'A'): -1, ('Y', 'W'): 2, ('E', 'E'): 5,
('Y', 'S'): -2, ('C', 'N'): -3, ('V', 'C'): -1, ('T', 'H'): -2,
('P', 'R'): -2, ('V', 'G'): -3, ('T', 'L'): -1, ('V', 'K'): -2,
('K', 'Q'): 1, ('R', 'A'): -1, ('I', 'R'): -3, ('T', 'D'): -1,
('P', 'F'): -4, ('I', 'N'): -3, ('K', 'I'): -3, ('M', 'D'): -3,
('V', 'W'): -3, ('W', 'W'): 11, ('M', 'H'): -2, ('P', 'N'): -2,
('K', 'A'): -1, ('M', 'L'): 2, ('K', 'E'): 1, ('Z', 'E'): 4,
('X', 'N'): -1, ('Z', 'A'): -1, ('Z', 'M'): -1, ('X', 'F'): -1,
('K', 'C'): -3, ('B', 'Q'): 0, ('X', 'B'): -1, ('B', 'M'): -3,
('F', 'C'): -2, ('Z', 'Q'): 3, ('X', 'Z'): -1, ('F', 'G'): -3,
('B', 'E'): 1, ('X', 'V'): -1, ('F', 'K'): -3, ('B', 'A'): -2,
('X', 'R'): -1, ('D', 'D'): 6, ('W', 'G'): -2, ('Z', 'F'): -3,
('S', 'Q'): 0, ('W', 'C'): -2, ('W', 'K'): -3, ('H', 'Q'): 0,
('L', 'C'): -1, ('W', 'N'): -4, ('S', 'A'): 1, ('L', 'G'): -4,
('W', 'S'): -3, ('S', 'E'): 0, ('H', 'E'): 0, ('S', 'I'): -2,
('H', 'A'): -2, ('S', 'M'): -1, ('Y', 'L'): -1, ('Y', 'H'): 2,
('Y', 'D'): -3, ('E', 'R'): 0, ('X', 'P'): -2, ('G', 'G'): 6,
('G', 'C'): -3, ('E', 'N'): 0, ('Y', 'T'): -2, ('Y', 'P'): -3,
('T', 'K'): -1, ('A', 'A'): 4, ('P', 'Q'): -1, ('T', 'C'): -1,
('V', 'H'): -3, ('T', 'G'): -2, ('I', 'Q'): -3, ('Z', 'T'): -1,
('C', 'R'): -3, ('V', 'P'): -2, ('P', 'E'): -1, ('M', 'C'): -1,
('K', 'N'): 0, ('I', 'I'): 4, ('P', 'A'): -1, ('M', 'G'): -3,
('T', 'S'): 1, ('I', 'E'): -3, ('P', 'M'): -2, ('M', 'K'): -1,
('I', 'A'): -1, ('P', 'I'): -3, ('R', 'R'): 5, ('X', 'M'): -1,
('L', 'I'): 2, ('X', 'I'): -1, ('Z', 'B'): 1, ('X', 'E'): -1,
('Z', 'N'): 0, ('X', 'A'): 0, ('B', 'R'): -1, ('B', 'N'): 3,
('F', 'D'): -3, ('X', 'Y'): -1, ('Z', 'R'): 0, ('F', 'H'): -1,
('B', 'F'): -3, ('F', 'L'): 0, ('X', 'Q'): -1, ('B', 'B'): 4
}
def local_alignment_with_affine_gap(str1, str2, gap_open=11, gap_extend=1):
m, n = len(str1), len(str2)
# Initialize score matrices
M = [[0] * (n + 1) for _ in range(m + 1)]
X = [[-float('inf')] * (n + 1) for _ in range(m + 1)]
Y = [[-float('inf')] * (n + 1) for _ in range(m + 1)]
# Initialize backtrack matrices
B = [[0] * (n + 1) for _ in range(m + 1)]
max_score, max_i, max_j = 0, 0, 0
# Fill matrices
for i in range(1, m + 1):
for j in range(1, n + 1):
X[i][j] = max(X[i][j-1] - gap_extend, M[i][j-1] - gap_open - gap_extend)
Y[i][j] = max(Y[i-1][j] - gap_extend, M[i-1][j] - gap_open - gap_extend)
key = (str1[i-1], str2[j-1]) if (str1[i-1], str2[j-1]) in BLOSUM62 else (str2[j-1], str1[i-1])
match_score = BLOSUM62[key]
M[i][j] = max(0, M[i-1][j-1] + match_score, X[i][j], Y[i][j])
if M[i][j] > max_score:
max_score, max_i, max_j = M[i][j], i, j
if M[i][j] == 0:
B[i][j] = 0
elif M[i][j] == M[i-1][j-1] + match_score:
B[i][j] = 1
elif M[i][j] == X[i][j]:
B[i][j] = 2
else:
B[i][j] = 3
# Backtrack
i, j = max_i, max_j
aligned_1, aligned_2 = [], []
while B[i][j] != 0:
if B[i][j] == 1:
aligned_1.append(str1[i-1])
aligned_2.append(str2[j-1])
i -= 1
j -= 1
elif B[i][j] == 2:
aligned_1.append('-')
aligned_2.append(str2[j-1])
j -= 1
else:
aligned_1.append(str1[i-1])
aligned_2.append('-')
i -= 1
return max_score, ''.join(reversed(aligned_1)), ''.join(reversed(aligned_2))
def parse_fasta(fasta_str):
sequences = {}
for record in fasta_str.strip().split('>')[1:]:
lines = record.split('\n')
sequences[lines[0]] = ''.join(lines[1:])
return list(sequences.values())
sample_input = """
>Rosalind_8
PLEASANTLY
>Rosalind_18
MEANLY
"""
stringA, stringB = parse_fasta(sample_input)
score, aligned_strA, aligned_strB = local_alignment_with_affine_gap(stringA, stringB)
print(score)
print(aligned_strA.replace("-", ""))
print(aligned_strB.replace("-", ""))- BLOSUM62 Matrix:
BLOSUM62is a dictionary containing scores for amino acid substitutions based on the BLOSUM62 matrix. It provides a scoring scheme for amino acid matches and mismatches.
local_alignment_with_affine_gapFunction:- Inputs:
str1andstr2(the sequences to align),gap_openandgap_extend(penalties for opening and extending gaps). - Initialization:
M,X, andYare matrices used to store scores for alignments and gaps.Bis a backtracking matrix to reconstruct the optimal alignment.
- Matrix Filling:
- Iterates over each position in the matrices, computing scores based on the BLOSUM62 matrix and gap penalties.
- Updates the matrices to reflect the best alignment score at each position.
- Backtracking:
- Constructs the optimal local alignment by following the backtracking matrix.
- Inputs:
parse_fastaFunction:- Input: A string in FASTA format containing sequences.
- Output: A list of sequences parsed from the FASTA format.
- Example Usage:
sample_input: A FASTA formatted string with two example sequences.- Execution:
- Parses the FASTA string into sequences.
- Performs local alignment on these sequences.
- Prints the alignment score and the aligned sequences (with gaps removed).
105 Identifying Reversing Substitutions
For a rooted tree \(T\) whose internal nodes are labeled with genetic strings, our goal is to identify reversing substitutions in \(T\). Assuming that all the strings of \(T\) have the same length, a reversing substitution is defined formally as two parent-child string pairs \((s,t)\) and \((v,w)\) along with a position index \(i\), where:
- there is a path in \(T\) from \(s\) down to \(w\);
- \(s[i]=w[i]≠v[i]=t[i\)]; and
- if \(u\) is on the path connecting \(t\) to \(v\), then \(t[i]=u[i]\).
In other words, the third condition demands that a reversing substitution must be contiguous: no other substitutions can appear between the initial and reversing substitution.
Given: A rooted binary tree \(T\) with labeled nodes in Newick format, followed by a collection of at most 100 DNA strings in FASTA format whose labels correspond to the labels of \(T\). We will assume that the DNA strings have the same length, which does not exceed 400 bp).
Return: A list of all reversing substitutions in \(T\) (in any order), with each substitution encoded by the following three items:
- the name of the species in which the symbol is first changed, followed by the name of the species in which it changes back to its original state
- the position in the string at which the reversing substitution occurs; and
- the reversing substitution in the form original_symbol->substituted_symbol->reverted_symbol.
105.1 Sample Dataset
(((ostrich,cat)rat,mouse)dog,elephant)robot;
>robot
AATTG
>dog
GGGCA
>mouse
AAGAC
>rat
GTTGT
>cat
GAGGC
>ostrich
GTGTC
>elephant
AATTC105.2 Sample Output
dog mouse 1 A->G->A
dog mouse 2 A->G->A
rat ostrich 3 G->T->G
rat cat 3 G->T->G
dog rat 3 T->G->T105.3 Solution
clas Node:
def __init__(self, number, parent, name=None):
self.number = number
self.parent = parent
self.children = []
self.name = name or f"Node_{number}"
def __repr__(self):
return f"Node_{self.number}({self.name})" if self.name != f"Node_{self.number}" else f"Node_{self.number}"
def add_child(self, child):
self.children.append(child)
clas Newick:
def __init__(self, data):
self.nodes = []
self.edges = []
self.construct_tree(data)
self.name_index = {node.name: node.number for node in self.nodes}
self.inv_name_index = {node.number: node.name for node in self.nodes}
def construct_tree(self, data):
tokens = data.replace(',', ' ').replace('(', '( ').replace(')', ' )').strip(';').split()
stack = [Node(-1, None)]
for token in tokens:
if token == '(':
new_node = Node(len(self.nodes), stack[-1].number)
self.nodes.append(new_node)
if len(self.nodes) > 1:
self.nodes[new_node.parent].add_child(new_node.number)
self.edges.append((new_node.parent, new_node.number))
stack.append(new_node)
elif token == ')':
stack.pop()
elif token.startswith(')'):
stack[-1].name = token[1:]
stack.pop()
else:
new_node = Node(len(self.nodes), stack[-1].number, token)
self.nodes.append(new_node)
self.nodes[new_node.parent].add_child(new_node.number)
self.edges.append((new_node.parent, new_node.number))
def traverse(self, node_index=0, order='pre'):
node = self.nodes[node_index]
if order == 'pre':
result = [node]
for child in node.children:
result.extend(self.traverse(child, order))
else: # post-order
result = []
for child in node.children:
result.extend(self.traverse(child, order))
result.append(node)
return result
def max_depth(self, node):
return max([self.max_depth(self.nodes[child]) for child in node.children], default=-1) + 1
def all_paths(self, node):
if not node.children:
return []
paths = []
stack = [(node, [node.name])]
while stack:
current, path = stack.pop()
for child_idx in current.children:
child = self.nodes[child_idx]
new_path = path + [child.name]
if len(new_path) >= 3:
paths.append(new_path)
stack.append((child, new_path))
return paths
def rsub(self, DNA_strings):
rsub_list = []
pre_order = self.traverse(order='pre')
k = len(next(iter(DNA_strings.values())))
for pos in range(k):
for node in pre_order:
if node.children:
for path in self.all_paths(node):
nucs = [DNA_strings[label][pos] for label in path]
if nucs[0] == nucs[-1] != nucs[1] and all(x == nucs[1] for x in nucs[1:-1]):
rsub_list.append([path[1], path[-1], str(pos + 1), "->".join([nucs[0], nucs[1], nucs[-1]])])
return rsub_list
def parse_fasta(lines):
sequences = {}
current_seq = []
current_name = ""
for line in lines:
line = line.strip()
if line.startswith(">"):
if current_name:
sequences[current_name] = "".join(current_seq)
current_name = line[1:]
current_seq = []
else:
current_seq.append(line)
if current_name:
sequences[current_name] = "".join(current_seq)
return sequences
sample_input = """
(((ostrich,cat)rat,mouse)dog,elephant)robot;
>robot
AATTG
>dog
GGGCA
>mouse
AAGAC
>rat
GTTGT
>cat
GAGGC
>ostrich
GTGTC
>elephant
AATTC
""".strip().split("\n")
newick = sample_input[0]
DNA_strings = parse_fasta(sample_input[1:])
tree = Newick(newick)
result = tree.rsub(DNA_strings)
for r in result:
print(" ".join(r))Here’s a refactored version of the provided code with explanations:
clas Node:
def __init__(self, number, parent, name=None):
self.number = number
self.parent = parent
self.children = []
self.name = name or f"Node_{number}"
def __repr__(self):
return f"Node_{self.number}({self.name})" if self.name != f"Node_{self.number}" else f"Node_{self.number}"
def add_child(self, child):
self.children.append(child)
clas Newick:
def __init__(self, data):
self.nodes = []
self.edges = []
self.construct_tree(data)
self.name_index = {node.name: node.number for node in self.nodes}
self.inv_name_index = {node.number: node.name for node in self.nodes}
def construct_tree(self, data):
tokens = data.replace(',', ' ').replace('(', '( ').replace(')', ' )').strip(';').split()
stack = [Node(-1, None)]
for token in tokens:
if token == '(':
new_node = Node(len(self.nodes), stack[-1].number)
self.nodes.append(new_node)
if len(self.nodes) > 1:
self.nodes[new_node.parent].add_child(new_node.number)
self.edges.append((new_node.parent, new_node.number))
stack.append(new_node)
elif token == ')':
stack.pop()
elif token.startswith(')'):
stack[-1].name = token[1:]
stack.pop()
else:
new_node = Node(len(self.nodes), stack[-1].number, token)
self.nodes.append(new_node)
self.nodes[new_node.parent].add_child(new_node.number)
self.edges.append((new_node.parent, new_node.number))
def traverse(self, node_index=0, order='pre'):
node = self.nodes[node_index]
if order == 'pre':
result = [node]
for child in node.children:
result.extend(self.traverse(child, order))
else: # post-order
result = []
for child in node.children:
result.extend(self.traverse(child, order))
result.append(node)
return result
def max_depth(self, node):
return max([self.max_depth(self.nodes[child]) for child in node.children], default=-1) + 1
def all_paths(self, node):
if not node.children:
return []
paths = []
stack = [(node, [node.name])]
while stack:
current, path = stack.pop()
for child_idx in current.children:
child = self.nodes[child_idx]
new_path = path + [child.name]
if len(new_path) >= 3:
paths.append(new_path)
stack.append((child, new_path))
return paths
def rsub(self, DNA_strings):
rsub_list = []
pre_order = self.traverse(order='pre')
k = len(next(iter(DNA_strings.values())))
for pos in range(k):
for node in pre_order:
if node.children:
for path in self.all_paths(node):
nucs = [DNA_strings[label][pos] for label in path]
if nucs[0] == nucs[-1] != nucs[1] and all(x == nucs[1] for x in nucs[1:-1]):
rsub_list.append([path[1], path[-1], str(pos + 1), "->".join([nucs[0], nucs[1], nucs[-1]])])
return rsub_list
def parse_fasta(lines):
sequences = {}
current_seq = []
current_name = ""
for line in lines:
line = line.strip()
if line.startswith(">"):
if current_name:
sequences[current_name] = "".join(current_seq)
current_name = line[1:]
current_seq = []
else:
current_seq.append(line)
if current_name:
sequences[current_name] = "".join(current_seq)
return sequences105.4 Classes and Their Functions
NodeClass:- Represents a single node in the tree.
- Each node has a unique number, a parent node, a list of children, and a name.
NewickClass:- Purpose: Parses and builds a tree from a Newick format string.
- Key Methods:
construct_tree(data): Builds the tree structure from the Newick format string.traverse(): Gets all nodes in a specific order (pre-order or post-order).max_depth(node): Finds the maximum depth of the tree from a given node.all_paths(node): Lists all paths starting from a node.rsub(DNA_strings): Finds specific patterns in the DNA sequences based on the tree.
parse_fasta(lines):
- Converts FASTA formatted sequence data into a dictionary. Keys are sequence names, and values are the sequences.
105.5 How It Works
- Parse the Tree and Sequences:
- Newick Tree: Convert the Newick format string into a tree structure.
- FASTA Sequences: Read and store DNA sequences.
- Find Patterns:
- Use the tree structure and DNA sequences to find and list patterns where:
- The first and last characters of a pattern are the same.
- The middle characters are all the same but different from the first/last character.
- Use the tree structure and DNA sequences to find and list patterns where: